[Literature Review] Gene Knockout of ERAP1 Provides New Insights for Optimizing Tumor Immunotherapy
CRISPR/Cas9 gene knockout

Neuroblastoma (NB) is one of the most common pediatric tumors, typically occurring in the sympathetic nervous system of children, especially in the adrenal glands. Although some early-stage NB cases can be treated successfully through surgery and chemotherapy, most advanced cases exhibit resistance to conventional therapies due to the tumor’s growth characteristics, leading to suboptimal treatment outcomes.
Currently, immunotherapy has shown remarkable efficacy in adult cancers, with immune checkpoint inhibitors (ICI) being the most widely applied. However, NB cells tend to have low immunogenicity, lacking sufficient antigens to stimulate the immune system effectively, which makes these tumors less responsive to ICIs and other immunotherapies, posing a significant challenge in the field of immunotherapy.
To address this issue, researchers used CRISPR/Cas9 gene editing to knock out the Endoplasmic Reticulum Aminopeptidase 1 (ERAP1) gene in mouse tumor cells. ERAP1 trims antigenic peptides, aiding their binding to Major Histocompatibility Complex Class I (MHC I) molecules, thereby enhancing T cell recognition of tumor cells. Upon ERAP1 knockout, the cells generated new antigens, significantly increasing their immunogenicity.
To further validate this strategy, researchers introduced the Histone Deacetylase (HDAC) inhibitor Entinostat into the ERAP1-deficient cells and observed increased expression of MHC I and Programmed Death Ligand-1 (PD-L1), with enhanced tumor cell targeting by T cells.
The results indicate that the combination of ERAP1 knockout, Entinostat, and Programmed Death Protein-1 (PD-1) blockade significantly suppressed tumor growth and extended survival in mice, offering new possibilities for immunotherapy in non-immunogenic tumors.
This study was published in the Journal of Experimental & Clinical Cancer Research under the title “Combining ERAP1 silencing and entinostat therapy to overcome resistance to cancer immunotherapy in neuroblastoma.”

Original Article Link:https://doi.org/10.1186/s13046-024-03180-y
I. Downregulation of ERAP1 does not affect the surface expression of MHC class I molecules in 9464D cells
Firstly, researchers employed CRISPR/Cas9 gene editing technology, designing two single guide RNAs (sgRNAs) to target different exons of the ERAP1 gene in the murine neuroblastoma cell line 9464D, achieving ERAP1 knockout. The successful knockout was confirmed using Western blot and gene sequencing, showing complete suppression of ERAP1 protein expression.
To analyze ERAP1’s specific role in the antigen processing and presentation (APP) pathway, particularly its effect on surface MHC class I expression, the researchers treated both ERAP1-knockout and control cells with interferon-gamma (IFNγ), a molecule known to induce MHC I expression. Results showed no significant changes in MHC I expression in ERAP1-knockout 9464D cells, and both the knockout and control cells exhibited similar MHC I recovery levels following IFNγ treatment, indicating that ERAP1 deficiency does not affect IFNγ-induced MHC I expression.
Furthermore, by examining the expression levels of other APP pathway components (such as TAPBP and β2-microglobulin) and key components of the IFNγ signaling pathway (such as IRF1, IRF2, and STAT1), they found that ERAP1 knockout did not noticeably impact the expression of these components.
These findings suggest that, while ERAP1 plays a role in antigen processing, its absence does not significantly alter surface MHC I expression or interfere with the antigen presentation process, indicating that ERAP1 inhibition or knockout does not impair the immune system’s ability to recognize and target tumor cells.
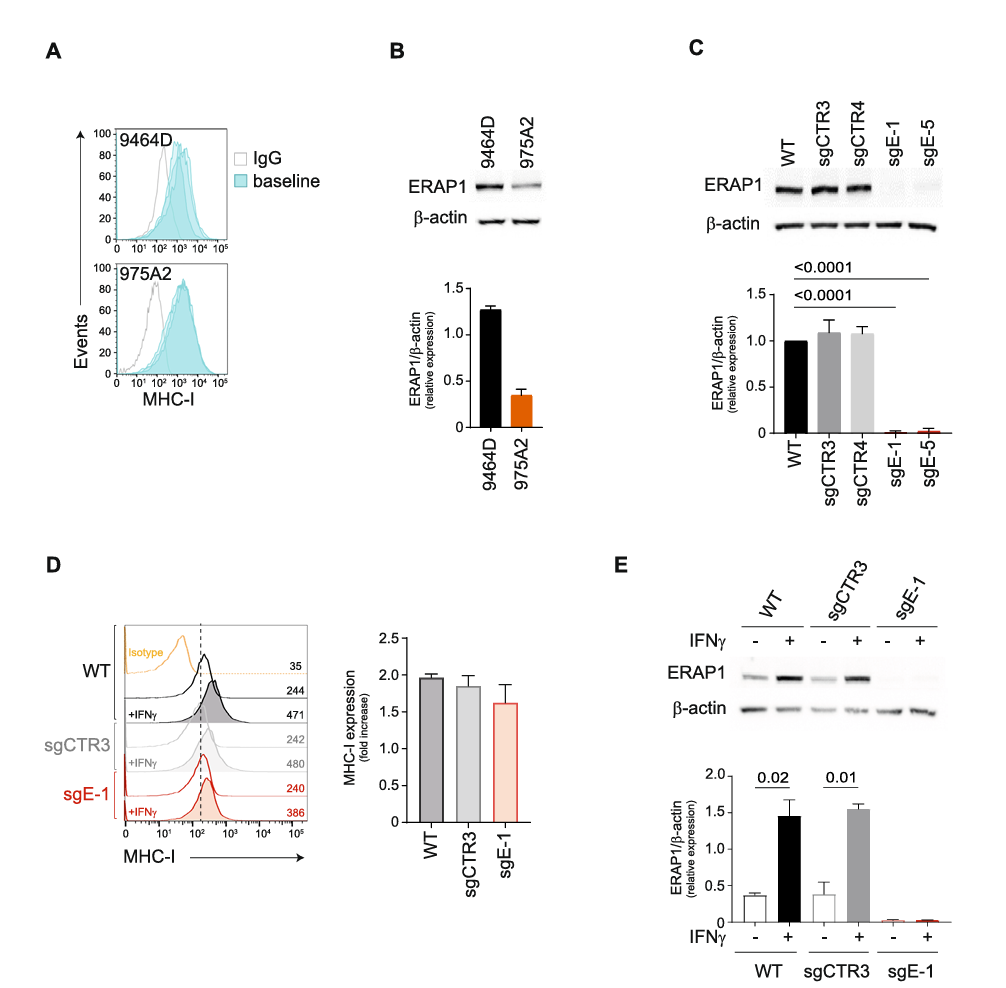
Fig. 1 Inhibition of ERAP1 does not affect the surface expression of MHC class I molecules of 9464D cells
II. Inhibition of ERAP1 renders 9464D cells more susceptible to lysis by immune cells
To investigate the effect of ERAP1 knockout on tumor cells, researchers named the ERAP1-knockout 9464D cell line as sgE-1 and created a control cell line, sgCTR3, by introducing a non-targeting sgRNA into 9464D cells. This ensured that the gene editing process was consistent between the experimental and control groups.
Previous studies have shown that ERAP1 silencing induces changes in the immunopeptidome, thereby affecting T cell- and NK cell-mediated anti-tumor responses. To evaluate the capacity of sgE-1 cells to activate immune cells, researchers co-cultured IFNγ-treated and untreated sgCTR3 and sgE-1 cells with splenocytes derived from wild-type 9464D tumor-bearing mice. Results indicated that, after 2 hours of co-culture, sgE-1 attracted significantly more CD3+ T cells, CD8+ T cells, NK cells, and NKT cells compared to sgCTR3.
Further protein array analysis of the supernatant from co-cultures of splenocytes and sgE-1 cells revealed that, compared to sgCTR3, immune cell attraction to relevant chemokines such as CX3CL1 and CCL5 increased more than twofold. Additionally, after 18 hours of co-culture, sgE-1 cells activated more CD8+ T cells and NK cells compared to control cells, with a significant increase in activation marker expression.
After 7 hours of co-culture, caspase 3/7-positive cells in sgE-1 increased, and by 24 hours, the number of live sgE-1 cells was significantly lower than that of sgCTR3. These data suggest that ERAP1 knockout promotes an increase in IFNγ release as well as the attraction and activation of T cells and NK cells, making tumor cells more susceptible to clearance by these immune cells.
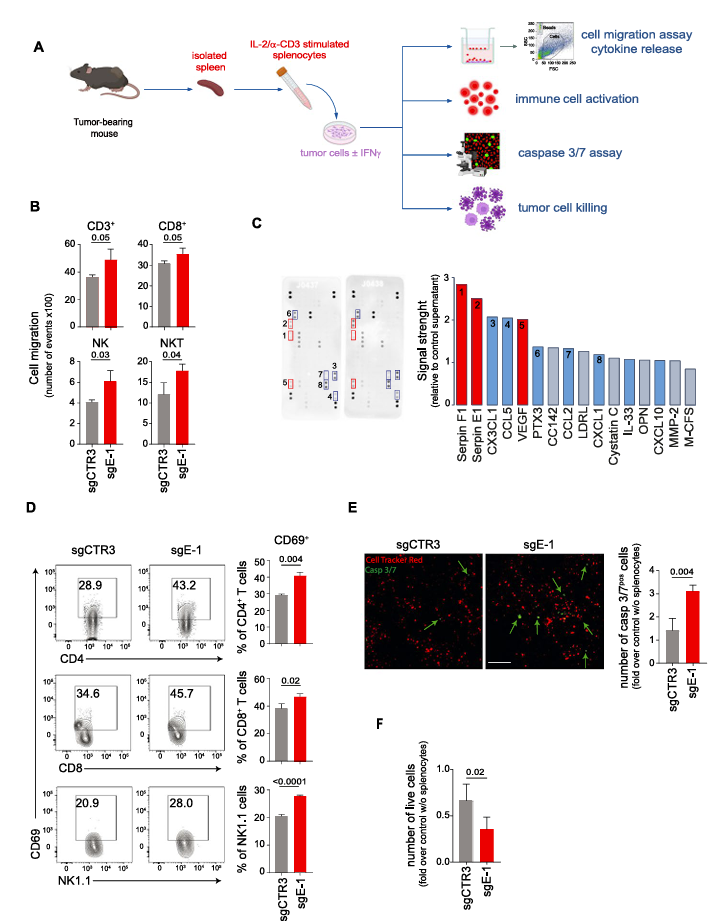
Fig. 2 Inhibition of ERAP1 renders 9464D cells more susceptible to lysis by immune cells
III. Lack of ERAP1 is not sufficient to control the in vivo growth of 9464D cells
Researchers implanted ERAP1-knockout sgE-1 cells and control sgCTR3 cells into the left flank of syngeneic C57BL/6 mice to observe tumor growth and monitor tumor volume changes over time. Results showed that although ERAP1 deficiency enhanced immune cell-mediated tumor targeting in vitro, it was insufficient to effectively control the growth of 9464D tumors in vivo.
A multi-color flow cytometry analysis of different immune cell populations in the tumor tissue revealed no differences in immune cell infiltration. This phenomenon may be due to the low level of MHC class I expression in 9464D tumors, which likely limited the immune response triggered by ERAP1 knockout and consequently reduced its inhibitory effect on tumor growth in vivo.
These findings suggest that ERAP1 knockout alone may not be sufficient to induce a robust immune response to control neuroblastoma growth, and additional strategies may be required to further enhance the tumor’s immunogenicity.
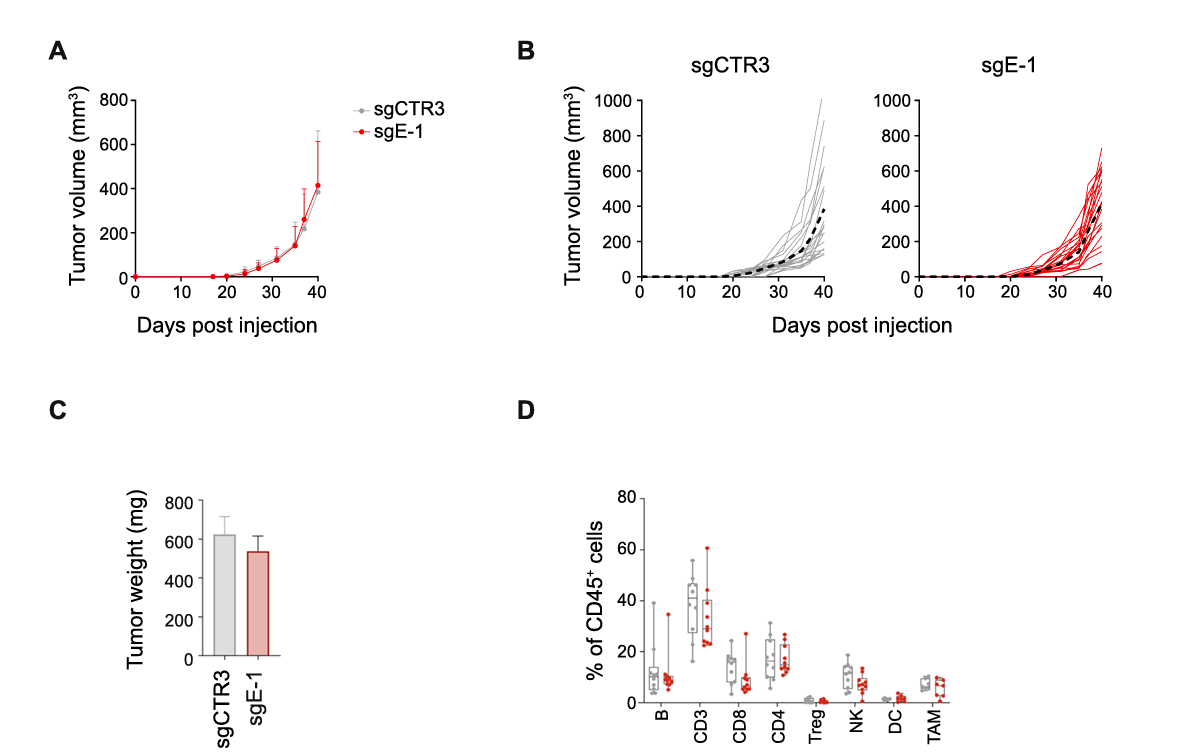
Fig. 3 ERAP1 inhibition does not alter the growth and immune landscape of 9464D tumors
IV. The lack of ERAP1 affects the expression of entinostat‑induced MHC class I molecules in 9464D cells
By treating 9464D cells with different concentrations of entinostat, researchers found that a 2μM concentration of entinostat over 48 hours increased surface expression of MHC class I molecules by at least twofold while maintaining cell viability. Further results showed that entinostat induced MHC class I expression in both control sgCTR3 and knockout sgE-1 cells, but the expression level was significantly reduced in sgE-1 cells lacking ERAP1.
Additionally, the expression of the non-classical MHC class I molecule Qa-1b was also induced in both sgCTR3 and sgE-1 cells, although at a lower level in sgE-1 cells, suggesting that ERAP1 may play a role in the generation of Qa-1b ligands. In contrast, molecules that do not bind peptides, such as RAE1 and CD86, were induced similarly in both sgCTR3 and sgE-1 cells, indicating that the role of ERAP1 in MHC class I molecules is specific.
To further explore the impact of ERAP1 on the immunopeptidome, researchers performed mass spectrometry analysis on entinostat-treated sgE-1 and sgCTR3 cells. Peptides were purified and eluted from H-2Kb and H-2Db molecules, identifying 3419 peptides (8–14 amino acids) and 7776 peptides (9–14 amino acids), respectively.
Using the NetMHC-4.0 tool to predict the binding affinity of these peptides to H-2Kb and H-2Db, the analysis showed that typical peptide lengths for H-2Kb and H-2Db were significantly reduced in sgE-1 cells, while longer peptides increased. However, this difference was not observed in peptides shared between sgCTR3 and sgE-1 cells, further supporting the conclusion that ERAP1 deficiency generates a unique immunopeptidome.
Additionally, an analysis of conserved amino acid distributions within peptide sequences revealed that ERAP1 deficiency affects the composition of typical 8-mers and 9-mers bound to H-2Kb and H-2Db, respectively. Notably, the conserved amino acid motif at the P5 position in H-2Kb-bound peptides was entirely lost.
These findings indicate that the canonical length and composition of the immunopeptidome presented by H-2Kb and H-2Db are highly dependent on ERAP1, resulting in a unique immunopeptidome in ERAP1-deficient cells.
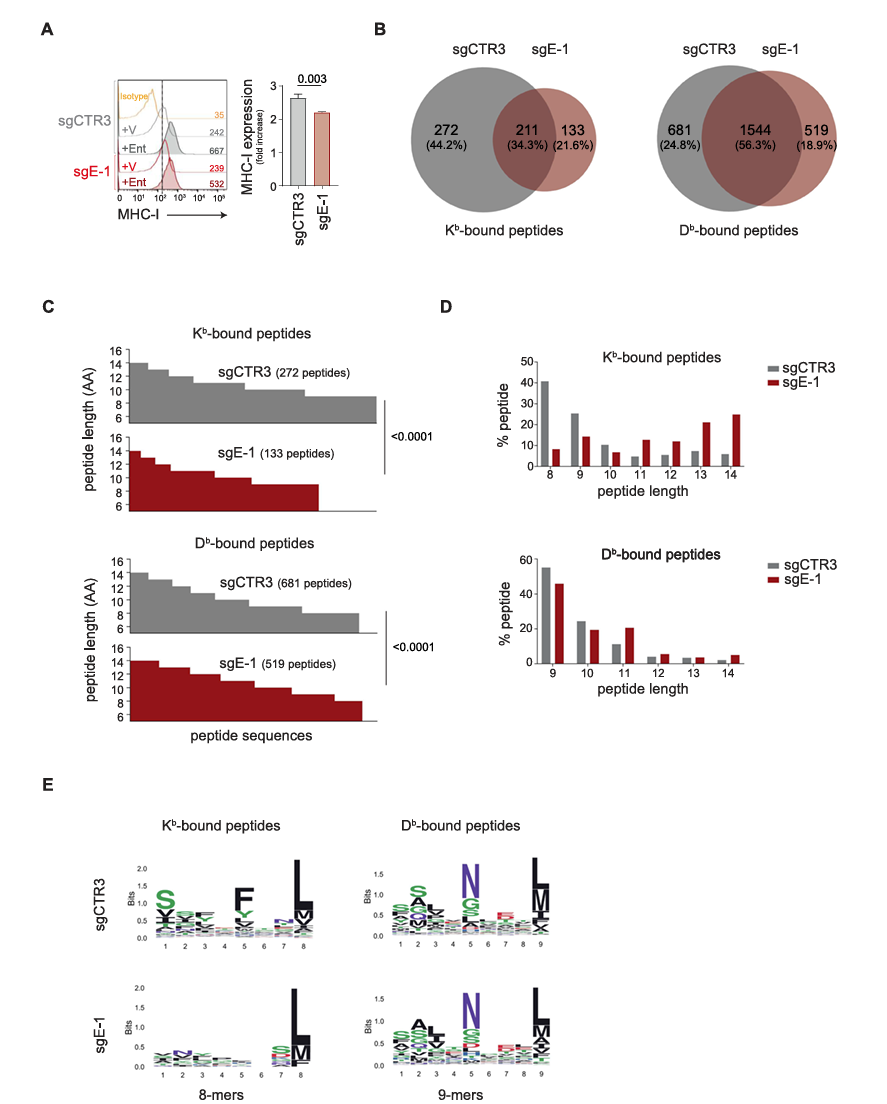
Fig. 4 Inhibition of ERAP1 affects both the surface expression of MHC class I molecules and the immunopeptidome of 9464D cells treated with entinostat
V. Lack of ERAP1 during entinostat treatment reprograms the tumor immune microenvironment to delay tumor progression
To evaluate the in vivo antitumor effect of entinostat combined with ERAP1 silencing, researchers subcutaneously injected sgCTR3 and sgE-1 cells into the flanks of C57BL/6 mice. Results showed that entinostat significantly inhibited the progression of sgE-1 tumors but had no significant effect on sgCTR3 tumors.
The slowed tumor progression also significantly extended host survival; at 46 days post-injection, all control mice had died, while 60% of mice bearing sgE-1 tumors remained alive, indicating that ERAP1 deficiency rendered 9464D tumors responsive to entinostat treatment. Consistent with in vitro findings, entinostat treatment significantly increased the surface expression of MHC class I molecules.
Additionally, entinostat shifted NB cells from the adrenergic (ADR) phenotype to a more immunogenic mesenchymal (MES) phenotype. Immunohistochemical analysis showed elevated expression of the MES marker SOX9 in entinostat-treated tumors, independent of ERAP1 expression.
Moreover, analysis of various immune cell subsets in tumors after 10 days of treatment revealed significantly increased immune infiltration in entinostat-treated sgE-1 tumors, particularly in tumor-infiltrating lymphocytes (TILs), CD8+ T cells, and CD4+ T cell subsets. Although the increase in NK cells was not significant, overall immune cell infiltration was enhanced. Immunofluorescence analysis confirmed these results, showing a notable increase in CD8+ T cells expressing IFNγ and granzyme B, as well as NK cells expressing IFNγ in entinostat-treated sgE-1 tumors.
These findings suggest that ERAP1 deficiency combined with entinostat treatment promotes the formation of an inflammatory T cell phenotype, thereby achieving control over neuroblastoma (NB) growth.
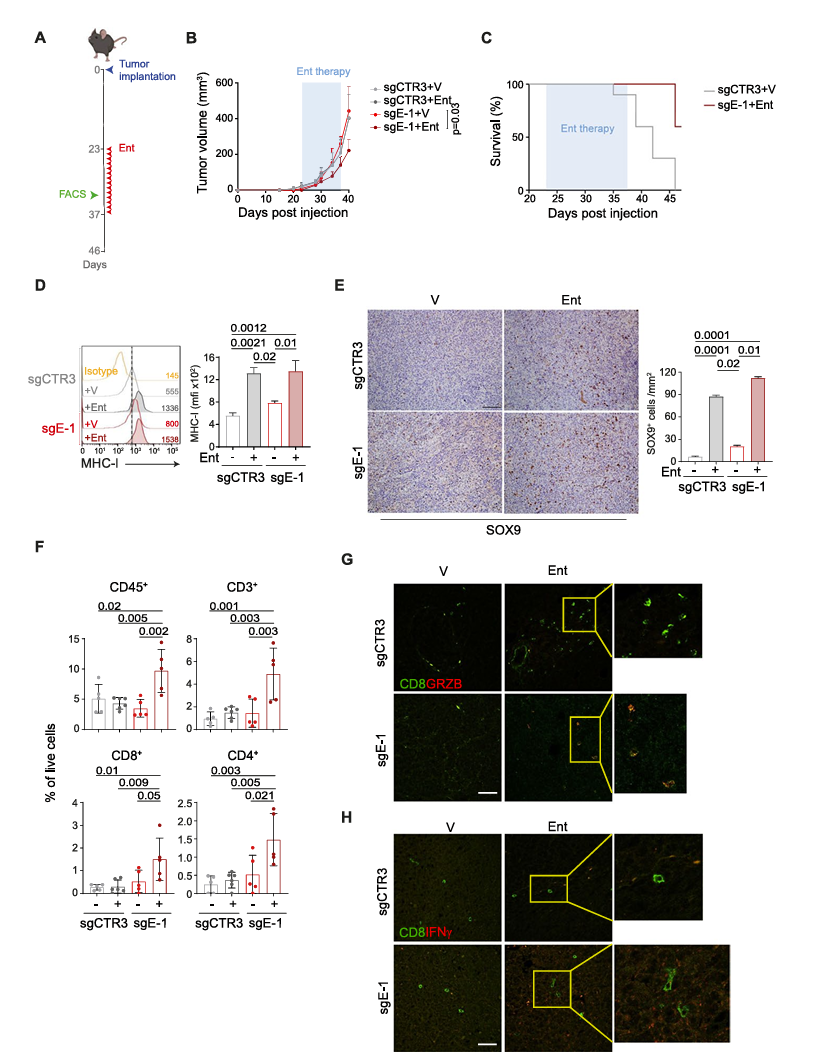
Fig. 5 Inhibition of ERAP1 in combination with entinostat treatment delays the growth of 9464D tumors and reshapes the intratumoral immune infiltrate
VI. Lack of ERAP1 in combination with entinostat and PD‑1 blockade control tumor growth and increase host survival
Previous studies have shown that entinostat can induce PD-L1 expression in various tumor models. In this study, entinostat-treated 9464D cells exhibited increased PD-L1 expression both in vitro and in vivo, independent of ERAP1 expression levels. To test whether entinostat could enhance the efficacy of PD-1 blockade, researchers injected sgE-1 and sgCTR3 cells into C57BL/6 mice. Once the tumors grew to 100 mm³, the mice were divided into four groups: control, entinostat alone, PD-1 blockade alone, and the combination of entinostat and PD-1 blockade.
The results showed that entinostat was significantly more effective in ERAP1-deficient tumors, while PD-1 blockade alone had no noticeable effect regardless of ERAP1 status. Analysis of survival revealed that the combination of entinostat and PD-1 blockade was more effective in sgE-1 tumors compared to other treatments.
In sgCTR3 tumor-bearing mice, the combined treatment had a similar effect to entinostat alone, with 40% survival. In contrast, in sgE-1 tumor-bearing mice, combination therapy significantly improved survival; all mice with sgE-1 tumors receiving combination treatment survived beyond 46 days, whereas only 57% of those treated with entinostat alone survived.
Additionally, immunohistochemistry analysis revealed a marked increase in CD8+ T cell infiltration in sgE-1 tumors treated with the combination therapy. These results suggest that ERAP1 deficiency plays a critical role in enhancing the anti-tumor effects of entinostat combined with PD-1 blockade, converting the previously unresponsive 9464D tumor model to a responsive state for PD-1 blockade therapy.
Conclusion
Overall, these findings indicate that ERAP1 plays a crucial role in tumor immune regulation, and its knockout can improve the responsiveness of neuroblastoma to immunotherapy. In addition, ERAP1 inhibition can increase tumor immunogenicity by upregulating the expression of MHC class I molecules and non-classical MHC molecules, while also working synergistically with entinostat and PD-1 blockade to effectively slow tumor growth and significantly extend host survival.
These discoveries provide new strategies for immunotherapy in non-immunogenic tumors, such as neuroblastoma, suggesting that ERAP1 may be a potential target in future cancer immunotherapy, as modulating its activity could enhance tumor sensitivity and therapeutic efficacy in immunotherapy.
EDITGENE has innovatively developed an efficient gene knock-out technology, offering customized gene knock-out services using an upgraded CRISPR/Cas9 system. With over a decade of experience in gene editing, EDITGENE has refined the optimal gRNA and homology arm design strategies, ensuring that their gene knock-out services deliver higher positive rates and broader applicability across various gene loci!!
Recent Blogs
1.[Weekly News]CRISPR/Cas9 Gene Knockout: A New Strategy to Overcome Immune Therapy Resistance in Neuroblastoma
2.[Literature Review] CRISPR Detection Novel Breakthrough: Harnessing CrisprAIE for Accurate and Rapid Detection
3.[Weekly News] CRISPR/Cas9 Screening Unlocks Secrets of Stem Cell Power
Follow us on social media





Contact us
+ 833-226-3234 (USA Toll-free)
+1-224-345-1927 (USA)
info@editxor.com
Tag
Share




![[Literature Review] Gene Knockout of ERAP1 Provides New Insights for Optimizing Tumor Immunotherapy](/uploads/20241111/eI5tQ8ChZSgOEkmw_53c82bdd67704fe0e159246934f924ee.png)

Comment (4)