[Literature Review] Cyclin D1 Therapy Leaves Cancer Cells Nowhere to Hide
Gene Editing
Overexpress cell line

Mantle cell lymphoma (MCL) is an aggressive B-cell–derived hematologic malignancy, characterized almost universally by oncogenic CCND1 aberrations leading to overexpression of cyclin D1 (Cyclin D1). Due to its poor response to conventional chemotherapy, high relapse rate, and lack of effective targeted therapies, the median overall survival of MCL patients is only 3–5 years, making the search for novel therapeutic strategies critical.
Recently, the research team led by Jithma Abeykoon published a study in the Journal of Clinical Investigation titled Cyclin D1 overexpression induces replication stress and microhomology-mediated end-joining dependence in mantle cell lymphoma. In this study, the researchers employed a Cyclin D1-overexpressing cell model and gene knockout technology to elucidate how Cyclin D1 overexpression induces replication stress, activates the POLθ-mediated microhomology-mediated end-joining (MMEJ) repair pathway, and to investigate the impact of deficiencies in the key DNA damage response kinase ATM on this process. These findings provide a theoretical foundation for targeting POLθ in the treatment of MCL.

Original link: https://doi.org/10.1172/JCI193006
Spotlight
1. Revealing a New Mechanism of Replication Stress
Demonstrated that Cyclin D1, acting as a transcription factor, directly activates the POLQ gene and selectively triggers the MMEJ repair pathway in hematologic malignancies, providing the first systematic elucidation of the replication stress mechanism in MCL.
2. Identifying a Novel Synthetic Lethal Target
Established Cyclin D1 overexpression as a biomarker for combined ATM/POLθ inhibition, and developed a novel “synthetic lethality” therapeutic strategy in cells with intact homologous recombination (HR).
3. Expanding the Therapeutic Scope of POLθ
Provided the first evidence of POLθ’s critical role in HR-proficient hematologic malignancies, uncovering its dual functions in both S phase and mitosis, thereby highlighting its broad therapeutic potential.
The researchers constructed a Cyclin D1 overexpression cell model in U2OS cells and examined markers of replication stress. The results showed that Cyclin D1 overexpression markedly increased p-RPA S33 foci (a classical marker of replication stress) and γ-H2AX foci (a marker of DNA double-strand breaks).
A standardized reporter gene system was then employed to assess changes in DNA repair pathway activity. This findings revealed that Cyclin D1 overexpression selectively activated the MMEJ pathway, indicating a specific reliance of cancer cells on MMEJ-mediated repair.
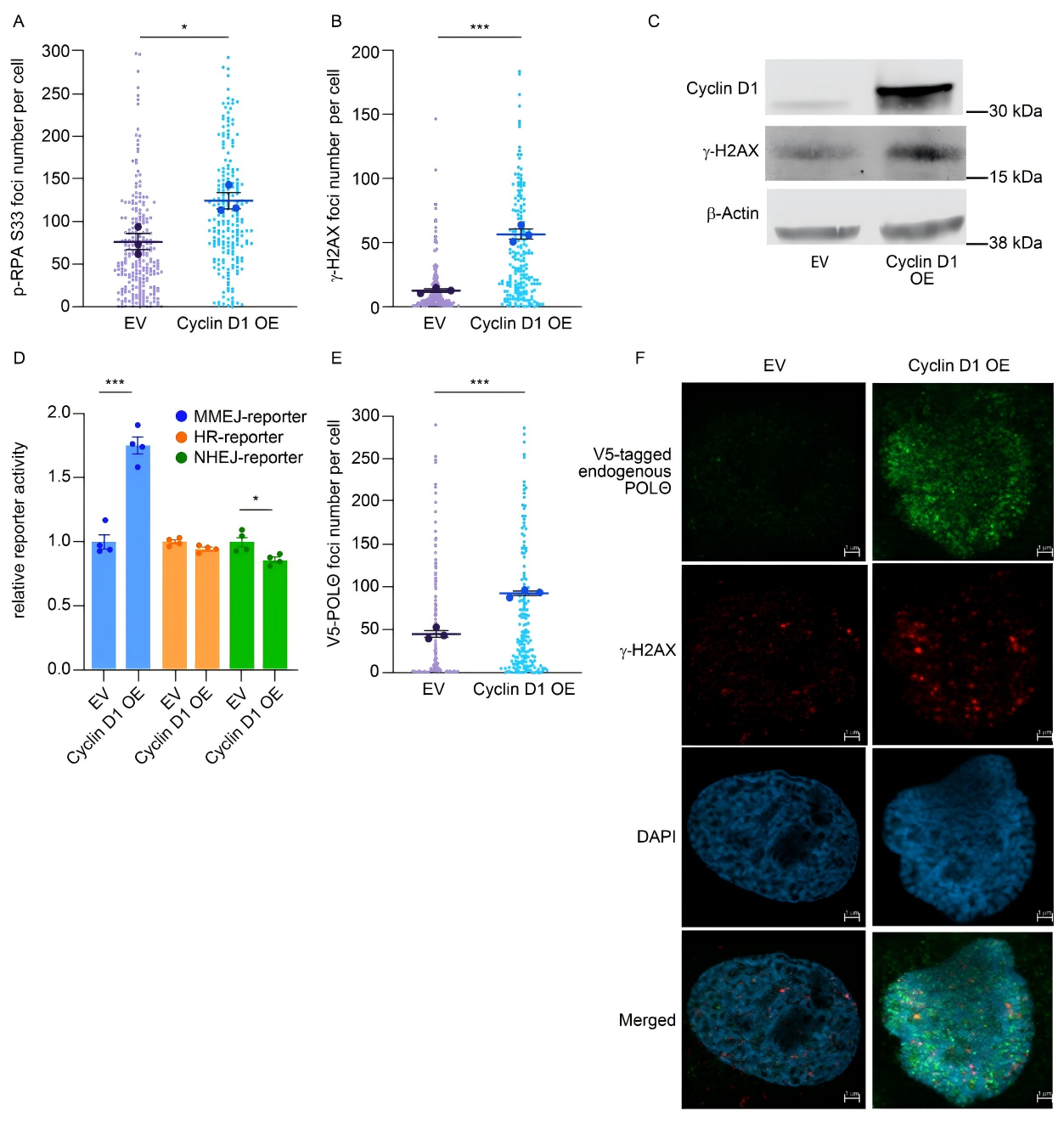
Figure 1. Cyclin D1 overexpression selectively activates MMEJ-mediated DNA damage repair
A POLQ knockout MCL cell line was generated using the CRISPR/Cas9 system and subsequently studied. The results showed that POLQ-deficient MCL cells were unable to effectively repair DNA damage during mitosis, accompanied by chromosomal abnormalities.
To elucidate the regulatory mechanism, the researchers employed techniques such as ChIP-qPCR and found increased enrichment of Cyclin D1 at the POLQ promoter region, along with enhanced promoter activity. These findings demonstrate that Cyclin D1 directly regulates POLQ transcription, thereby promoting MMEJ-mediated DNA damage repair.
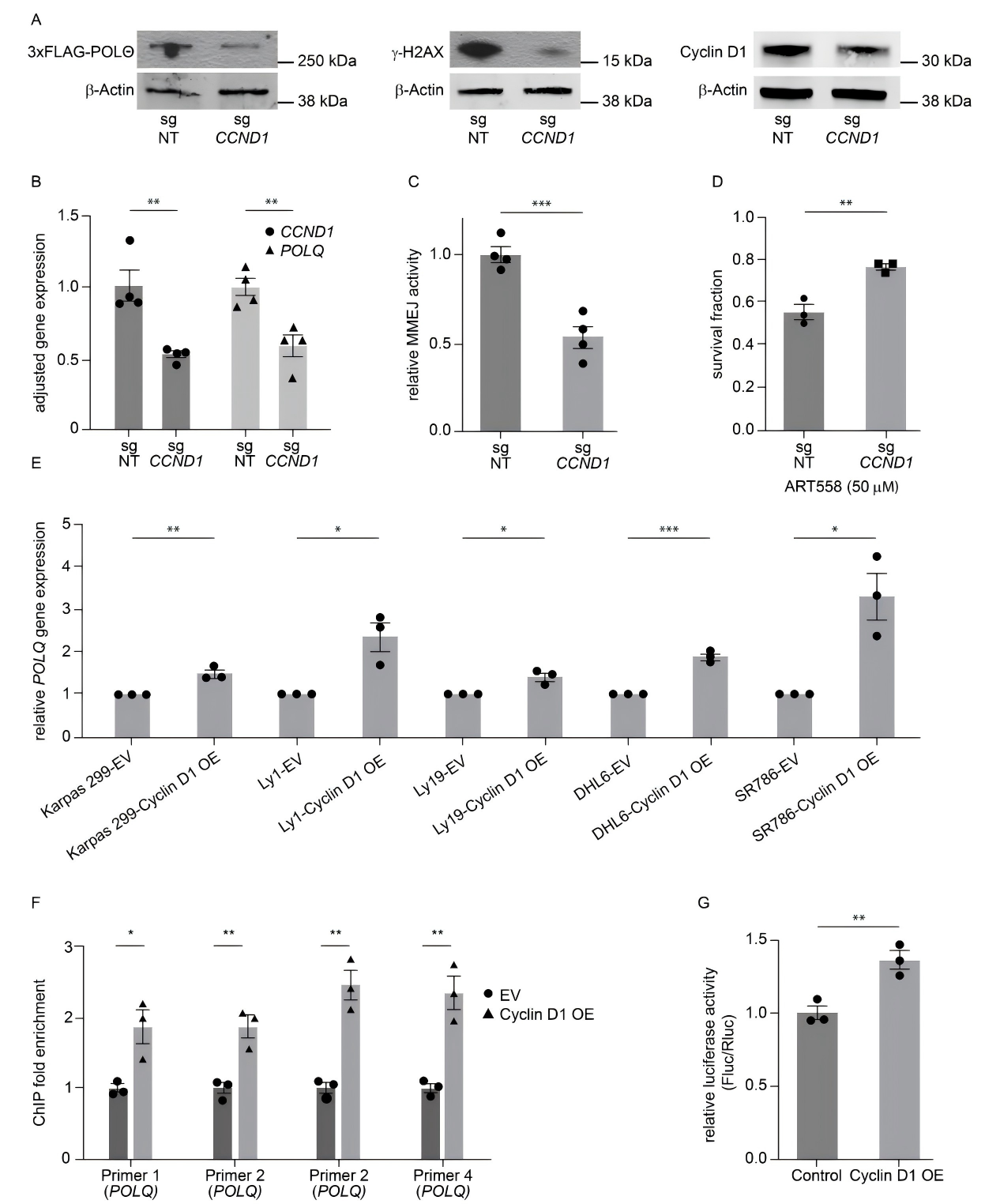
Figure 2. Cyclin D1 overexpression enhances DNA damage repair through specific interaction with POLQ
Given that 40%-50% of MCL patients harbor ATM gene defects, researchers generated ATM-deficient cell models to assess the impact. The experiments revealed that ATM deficiency further exacerbated replication stress in cyclin D1-overexpressing cells and markedly increased their reliance on the MMEJ pathway.
More importantly, POLQ expression was further upregulated in the context of ATM deficiency. This synergistic effect of dual defects provides a strong theoretical basis for therapeutic strategy development.
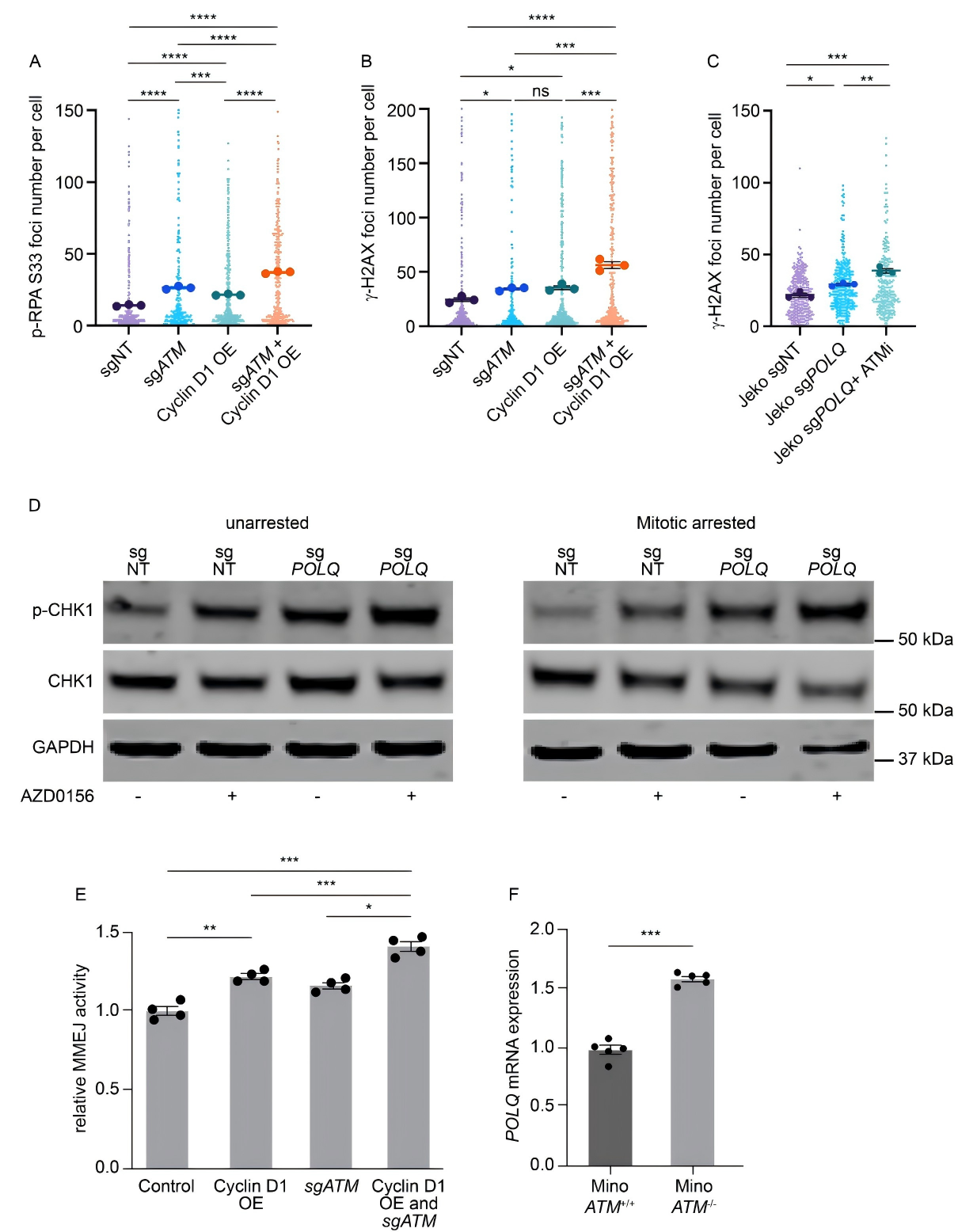
Figure 3. ATM deficiency and Cyclin D1 overexpression jointly enhance MMEJ-mediated DNA damage repair
The researchers treated MCL cell lines and primary patient samples with the POLθ inhibitors ART558 and novobiocin, and found that ATM-deficient MCL cells exhibited greater drug sensitivity.
In mouse models, POLθ inhibitors significantly suppressed tumor growth and prolonged survival, with minimal toxicity, providing strong experimental evidence to support clinical application in MCL, particularly for patients with ATM deficiency.
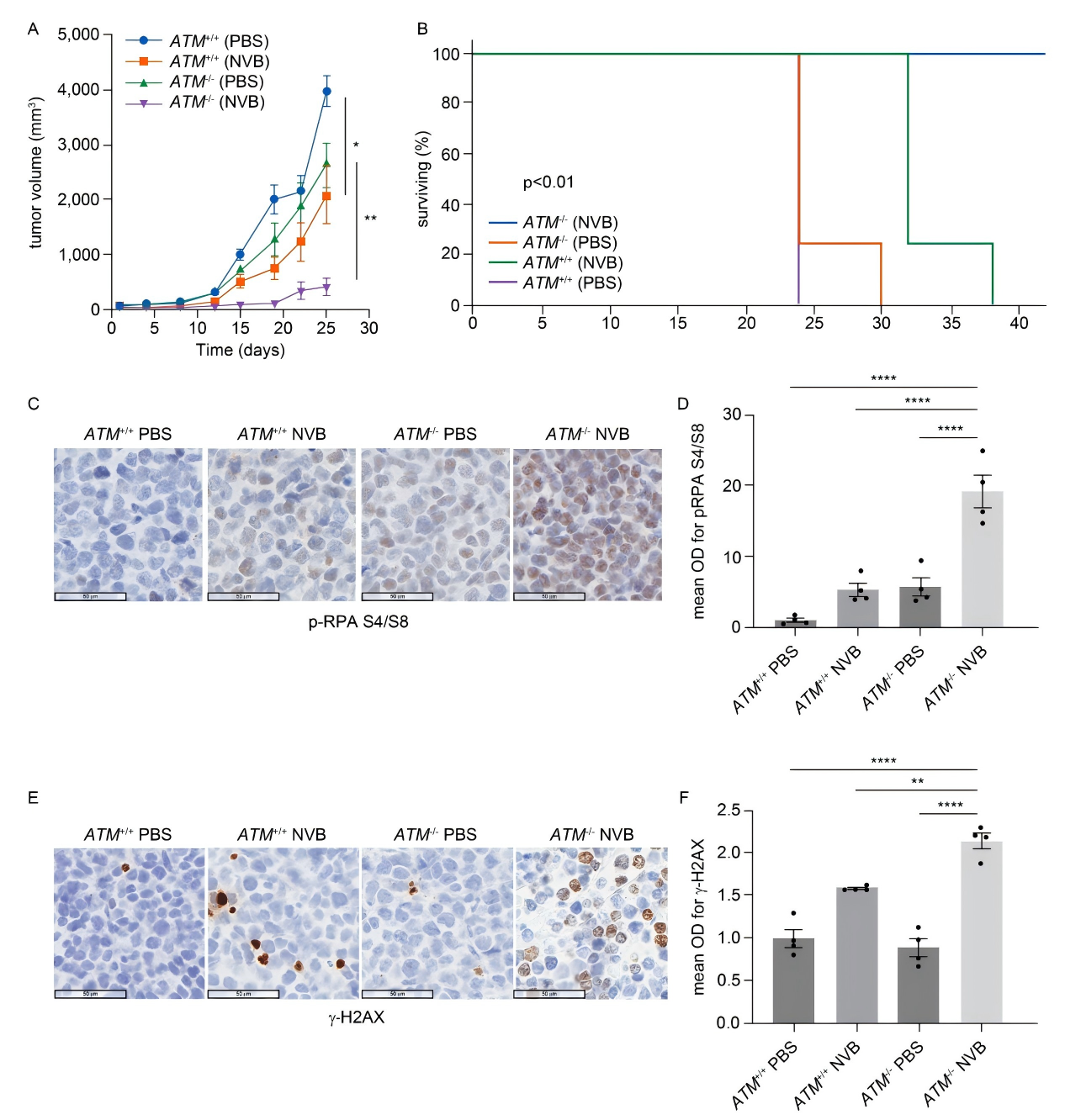
Figure 4. Antitumor effect of POLθ inhibition in an in vivo MCL model
This study elucidates the molecular mechanism by which cyclin D1 overexpression activates the MMEJ repair pathway and identifies POLθ as a key factor in maintaining genomic stability in MCL cells. These findings propose a novel therapeutic strategy for MCL and, given the widespread overexpression of cyclin D1 in various solid tumors, suggest that this mechanism may extend beyond MCL and be applicable to multiple solid tumor types.





Share







![[Literature Review] Cyclin D1 Therapy Leaves Cancer Cells Nowhere to Hide](/uploads/20250527/bL2GJjteMDvzmZys_53c82bdd67704fe0e159246934f924ee.png)

Comment (4)