




















[Literature Review] Targeting the USP37-HLTF Axis: A Promising Vulnerability in BRCA-Deficient Tumors
CRISPR screening

PARP inhibitors (PARPi) have significantly improved outcomes in BRCA-mutated tumors through synthetic lethality. However, resistance mechanisms—such as homologous recombination (HR) restoration or replication fork stabilization—lead to treatment failure in over 50% of patients.
A recent study published in Nucleic Acids Research, titled "USP37 counteracts HLTF to protect damaged replication forks and promote survival of BRCA1-deficient cells and PARP inhibitor resistance", sheds new light on this challenge.
Using genome-wide CRISPR screening , researchers identified the deubiquitinase USP37 as a key factor for BRCA1-deficient cell survival. For the first time, the study demonstrated that USP37 knockout cells exhibit a synthetic lethal effect in the context of BRCA1 deficiency. Moreover, USP37 was shown to bypass multiple resistance mechanisms, including loss of 53BP1, by stabilizing stalled replication forks.

Original link: DOI: 10.1093/nar/gkaf544
Spotlight
1.Novel Target Identification:
This study is the first to establish a synthetic lethal interaction between USP37 deficiency and BRCA1 loss, validated through double gene knockout models.
2.Breakthrough in Overcoming Clinical Resistance:
USP37 deficiency can reverse two major mechanisms of PARP inhibitor resistance—restored homologous recombination (e.g., 53BP1 loss) and stabilized replication forks.
3.Therapeutic Target Validation:
USP37 enzymatic activity is essential for cell survival under ATR inhibition, providing a clear direction for future drug development.
Using the TKOv3 lentiviral genome-wide CRISPR library screening, researchers conducted in a BRCA1-inducible depletion HeLa cell model under Olaparib (PARP inhibitor) treatment.
The results revealed that USP37 knockout reduced the survival of BRCA1-deficient cells by over 60%, and increased sensitivity to Olaparib by fourfold. Notably, even in the absence of PARPi treatment, USP37 loss alone was sufficient to induce significant cell death in BRCA1-depleted cells.
To validate this critical phenotype, a double knockout model—USP37 KO / BRCA1 mAID-HeLa—was constructed. The synthetic lethal effect was successfully confirmed and proven to be reproducible.
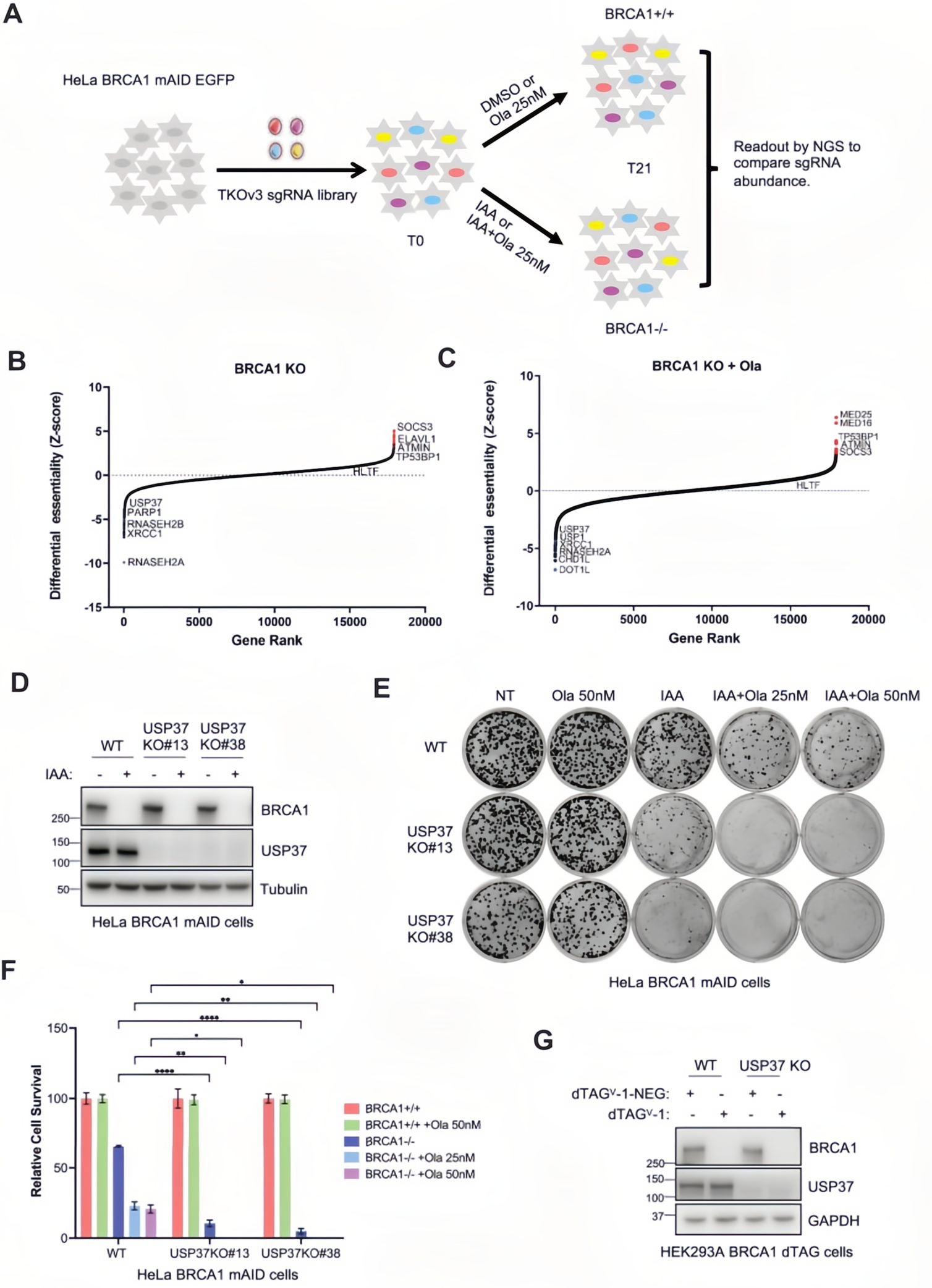
Figure 1. Impact of USP37 loss on cell viability in BRCA1-deficient cells
To investigate the molecular consequences of USP37 deficiency, the researchers applied DNA fiber analysis (IdU/CldU dual labeling) and hydroxyurea (HU)-induced replication stress.
In terms of DNA damage response, USP37 knockout in BRCA1-deficient cells led to a 3.5-fold increase in γH2AX foci, which further rose to a 5-fold increase following PARPi treatment. Micronucleus formation analysis revealed a dramatic rise—from 8% to 35%—in BRCA1-deficient cells lacking USP37.
Critically, DNA fiber assays assessing replication fork stability showed that BRCA1 deficiency alone reduced the CldU/IdU ratio by 40%, indicating nascent DNA degradation. With additional USP37 knockout, this ratio further dropped to 28%, confirming loss of replication fork protection. Importantly, RAD51 foci formation remained unchanged, ruling out the involvement of homologous recombination (HR) repair in this process.
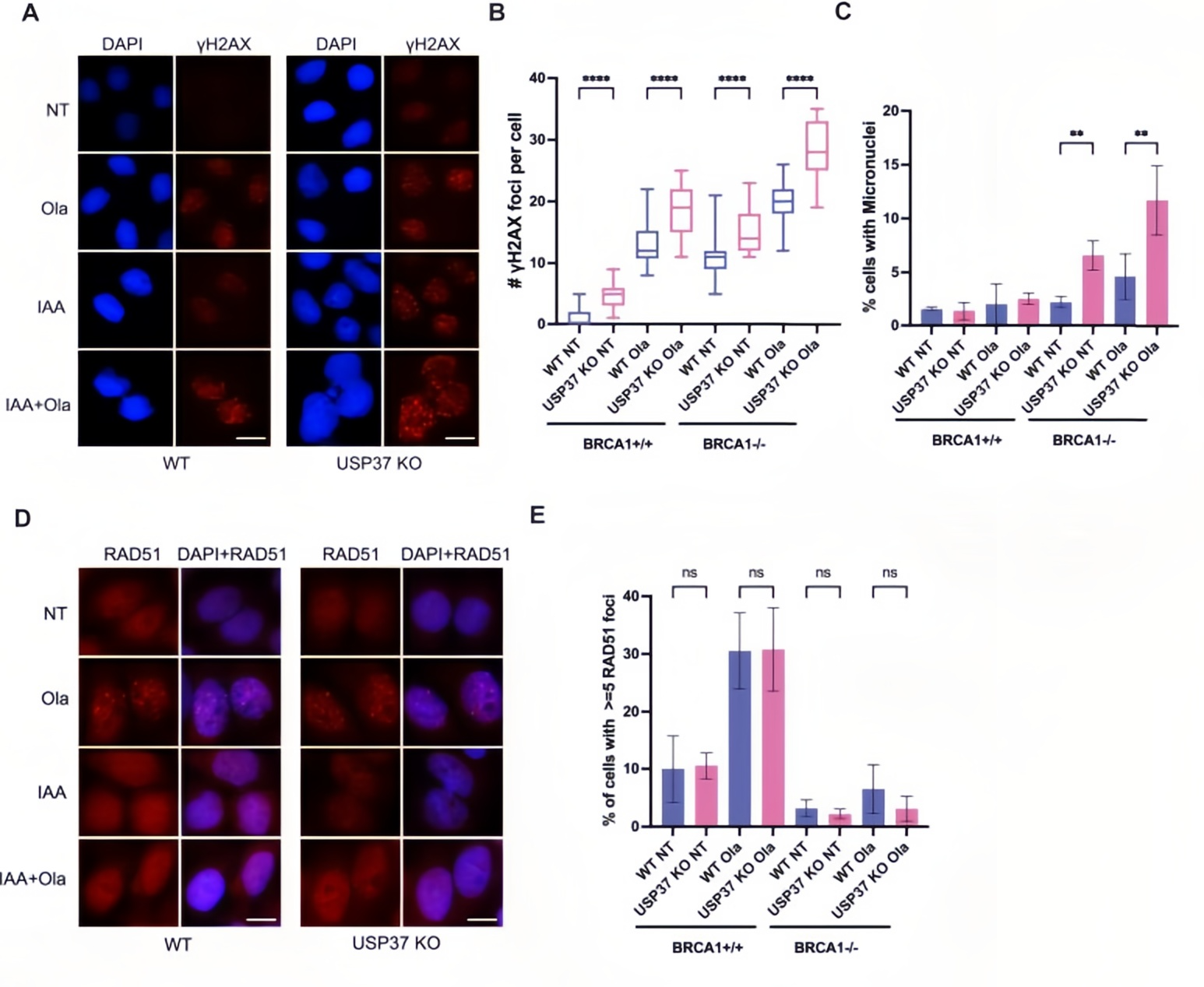
Figure 2. Effects of USP37 loss on genomic instability and replication fork stability in BRCA1-deficient cells
To uncover the molecular mechanism, the researchers performed co-immunoprecipitation (Co-IP) and in vivo deubiquitination assays. Co-IP experiments confirmed that Myc-tagged USP37 pulled down SFB-tagged RPA1, RPA2, and RPA3, indicating a direct interaction between USP37 and the RPA complex.
Functional assays showed that wild-type USP37 (WT) expression reduced RPA2-ubiquitin conjugate levels by 70%, while the catalytically inactive mutant USP37 C350S had no such effect. Correspondingly, RPA2 ubiquitination levels increased twofold in USP37 knockout cells.
Further enzymatic activity–dependent rescue experiments demonstrated that reintroducing wild-type USP37 restored resistance to ATR inhibitors (ATRi) and reduced chromatin-bound RPA levels. In contrast, the C350S mutant failed to rescue either phenotype.
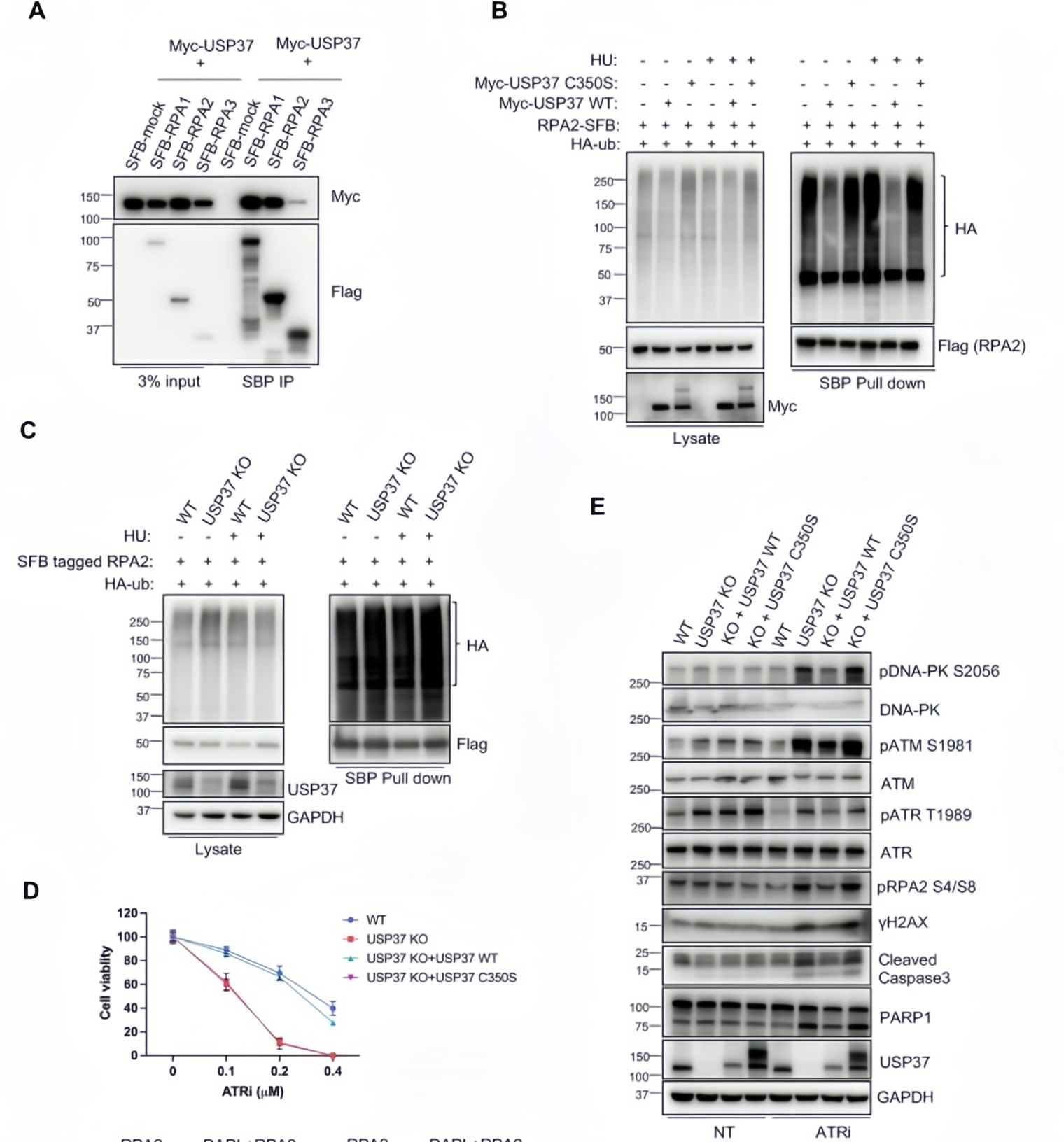
Figure 3. Functional validation of USP37 catalytic activity
To identify genetic interactions, the researchers conducted a secondary CRISPR screen in USP37 knockout cells, revealing HLTF as a key suppressor of the phenotype.
Mechanistically, BrdU immunofluorescence assays showed that USP37 knockout led to a threefold increase in HU-induced replication fork reversal (BrdU foci). Notably, this phenotype was rescued in USP37/HLTF double knockout (DKO) cells, with BrdU foci levels restored to those of wild-type controls. Functionally, HLTF knockdown improved the post-PARPi survival rate of BRCA1-deficient USP37 KO cells by 50%.
These findings suggest that USP37 maintains replication fork stability and prevents degradation by limiting HLTF accumulation at stalled forks, thereby reducing the incidence of double-strand breaks.
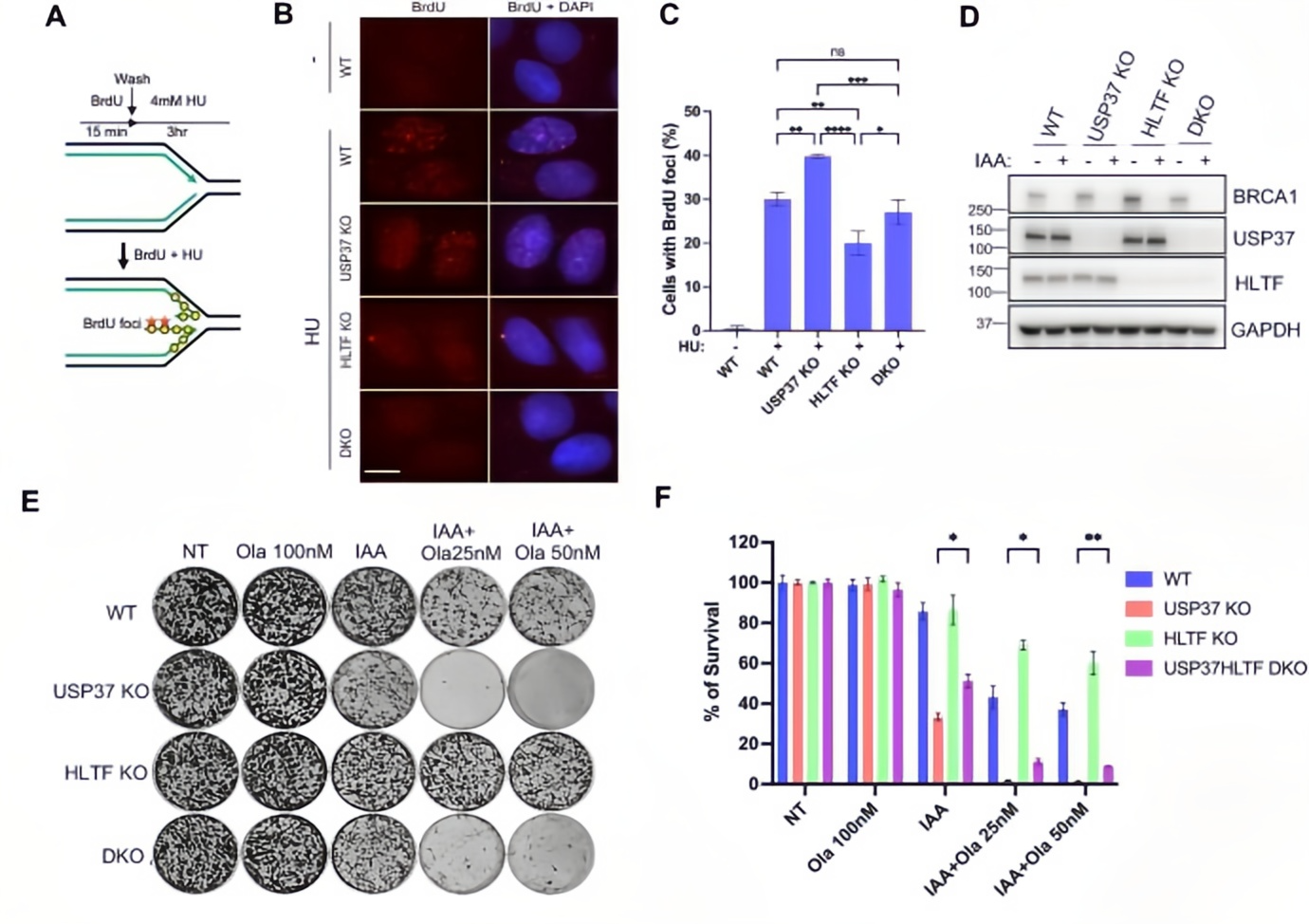
Figure 4. Rescue effects of HLTF loss on the USP37 knockout phenotype
This study provides in-depth insight into the critical role of USP37 in maintaining replication fork stability. It reveals, for the first time, a dual protective mechanism by which USP37 safeguards stalled replication forks: firstly, by deubiquitinating RPA to prevent excessive fork remodeling, and secondly, by antagonizing HLTF to inhibit replication fork reversal.
Further investigation into the chromatin-level interactions between USP37 and HLTF will help clarify the precise molecular regulation of DNA repair pathways. Such understanding may pave the way for novel therapeutic strategies targeting replication fork stability in cancer treatment.














