
 Login
Login
Alextina | Dec 28,2024
A talk about the Past and Present of Gene Editing Technology: ZFN, TALEN and CRISPR

Preface
Gene editing technology has always played a pivotal role in the exploration of life sciences. From the initial zinc finger nuclease (ZFN) to the current Prime Editing technology, gene editing has undergone a leapfrog development from concept to practice and from roughness to precision, providing powerful tools for us to understand and manipulate the essence of life.
Gene Mutation
When discussing gene editing, it is imperative to start with gene mutation. A gene point mutation, also known as a single base substitution, refers to a single-gene mutation where only one nucleotide pair is altered. This mutation can be either a transition, where a purine is replaced by another purine, a pyrimidine is replaced by another pyrimidine, or a transversion, which involves the substitution between a purine and a pyrimidine. Point mutations can occur spontaneously or be induced by mutagenic agents such as certain radiations and chemical agents. They can appear in coding or non-coding regions and may directly affect protein function or gene expression regulation, leading to synonymous mutations, missense mutations, nonsense mutations, or stop codon mutations. In coding regions, point mutations can result in amino acid substitutions, insertions, or deletions, thereby influencing protein structure and function. In non-coding regions, they may affect regulatory sequences of gene expression, such as promoters or enhancers, altering gene expression levels.
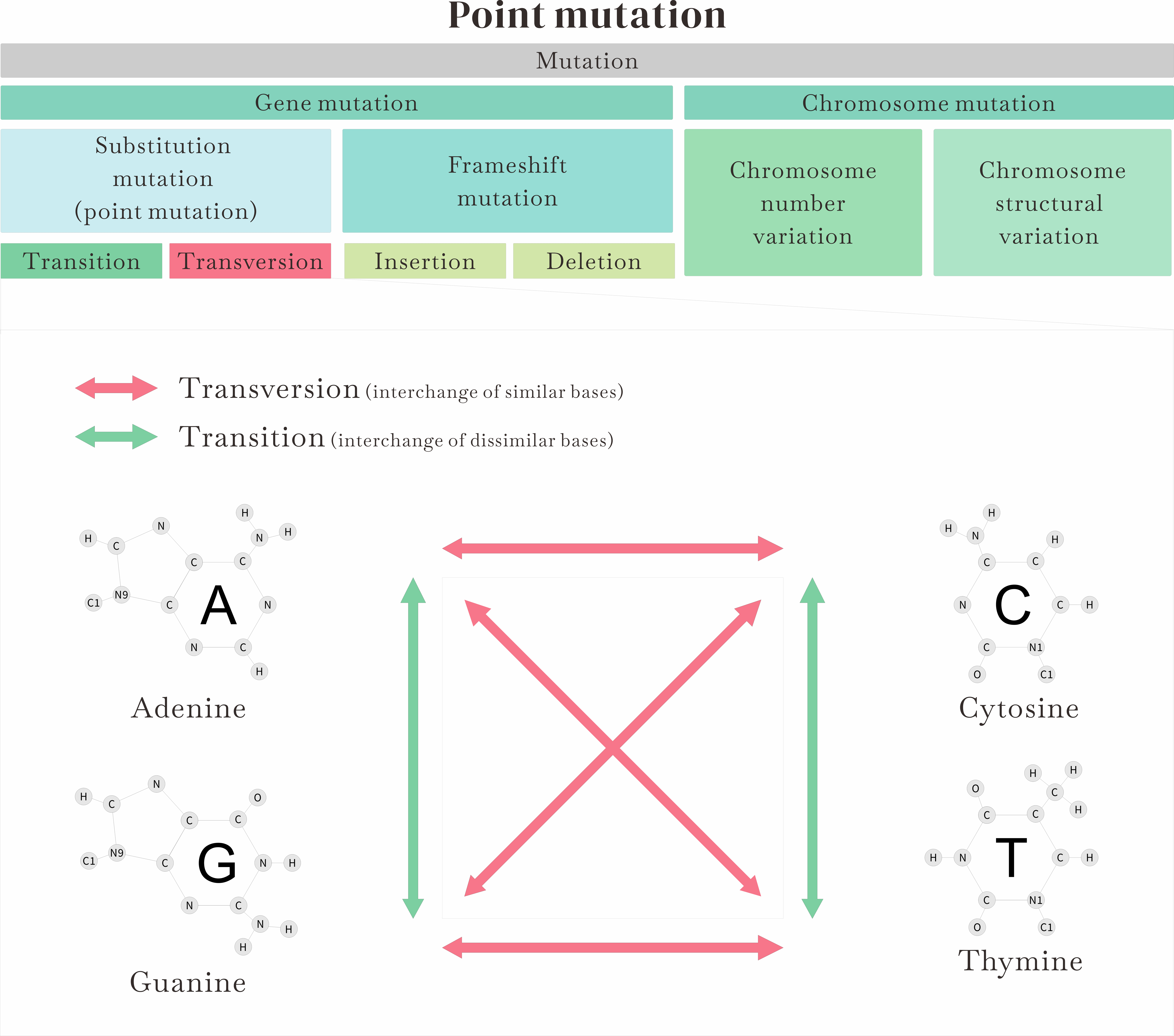
Humans are associated with over 75,000 diseases linked to genetic variations, with single-base mutations accounting for the largest proportion. Imagine if we could repair mutated genes back to their normal state; patients might be spared from the burden of rare diseases. Therefore, researchers are continuously exploring more precise gene editing tools to achieve this goal. We can study gene functions and their roles in organisms by precisely editing specific genes, such as base substitutions or gene knockouts. By altering specific bases in genes, we can influence the corresponding amino acid sequences and protein structures, elucidating the relationship between protein structure and function. Furthermore, studying the interaction between drugs and their targets provides powerful support for new drug development. It can also be used to repair disease-causing genes precisely, offering novel means for treating genetic diseases.
Next, let's review the fascinating development journey of gene editing technology.
Development History of Gene Editing Technologies
1. "The Golden Finger That Hits the Target Precisely" - Zinc Finger Nucleases (ZFNs)
Towards the end of the 1990s, the Aeron Klug laboratory discovered zinc finger proteins within the transcription factors of the African clawed frog. After artificial modification and attachment of a nucleic acid endonuclease, these proteins evolved into a genetic engineering editing tool known as zinc finger nuclease technology (ZFN).
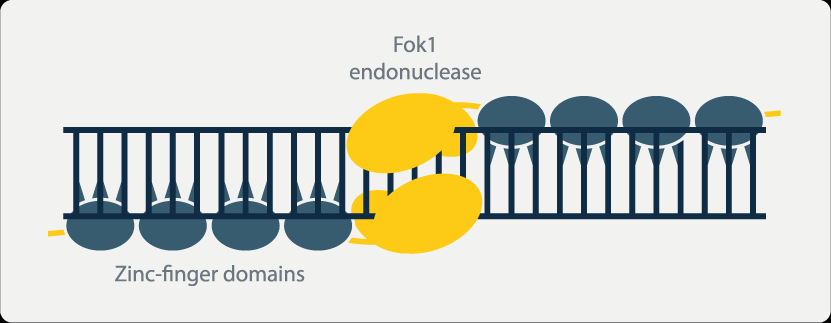
ZFNs rely on zinc-binding proteins to function, primarily used for site-specific editing and genomic modification. Zinc finger proteins are a class of proteins that possess the ability to bind to DNA. They contain structural domains that bind to zinc ions, known as zinc fingers. Each zinc finger typically recognizes and binds to 3 to 4 adjacent nucleotide base sequences. By combining different zinc fingers, it is possible to precisely design zinc finger proteins that specifically bind to target DNA sequences.
Moreover, when the nonspecific nucleic acid endonuclease FokI is linked to the zinc-binding protein, it acquires the ability to cleave DNA. Multiple zinc-binding proteins can be serially connected to form a zinc finger protein array. The interaction between these proteins results in enzymatic cleavage, creating double-strand breaks that stimulate the cell to initiate natural DNA repair processes. This triggers site-specific recombination, achieving the goal of gene editing.
However, ZFNs also have certain limitations. The individual zinc finger proteins within ZFNs can interact with each other, affecting their ability to recognize and bind to specific nucleotide sequences. This can lead to erroneous cleavage between two intended target fragments when multiple zinc finger proteins are serially connected, potentially causing off-target effects. Additionally, the target sequence length recognized by ZFNs is fixed, meaning that small genes and genes with high homology may not be effectively knocked out using this technology. Large gene fragments are also difficult to insert using ZFNs. Furthermore, the assembly process of ZFNs is non-modular, and the synthesis of zinc-binding proteins takes a long time. Difficulties in assembly in Escherichia coli, high costs, and patent barriers have restricted the widespread application of ZFN technology.
In November 2017, the first human clinical study evaluating the safety and tolerability of in vivo genomic editing therapy based on zinc finger nucleases (ZFN-based in vivo editing therapy) began administering the treatment to the first cohort of subjects. These three non-blinded, escalating single-dose Phase 1/2 studies, conducted for the first time in humans, aimed primarily to assess the safety and tolerability of ZFN-based in vivo editing therapy in Mucopolysaccharidosis Type I (MPS I) (n = 3), MPS II (n = 9), and Hemophilia B (n = 1). A total of 13 subjects were enrolled in the three studies: 9 subjects participated in the MPS II study, 3 subjects participated in the MPS I study, and 1 subject participated in the Hemophilia B study. Successful in vivo genomic editing by ZFN was demonstrated through the detection of albumin-IDS fusion mRNA in the livers of two MPS II subjects and albumin-IDUA fusion mRNA in the liver of one MPS I subject. At a dose of 1e13 vg/kg, MPS I subject 10 exhibited a transient increase in leukocyte iduronidase activity to half the normal level, providing evidence of in vivo genomic editing.

2. "The Mythical Nuclease" - TALEN
In 2007, German scientist Ulla Bonas discovered a sophisticated microinjection system in the plant bacterium Xanthomonas campestris. This bacterium injects a protein called AvrBs3 into plant cells, disguising itself as a plant transcription factor. This allows Xanthomonas campestris to synthesize proteins it urgently needs for its survival and reproduction. Subsequently, similar proteins were found in different bacteria and collectively named transcription activator-like effectors (TALEs).
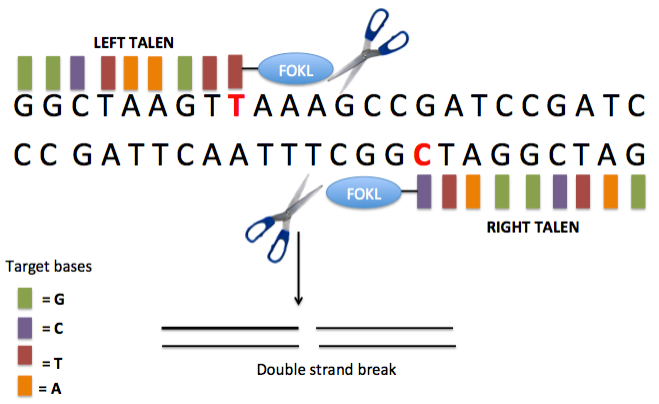
TALEN is a product of the combination of the DNA recognition and binding domain TALE with the nonspecific nucleic acid endonuclease FokI. Through dimerization, TALEN causes double-strand breaks in the target fragment, triggering the cell's NHEJ repair mechanism to repair the DNA damage. During the repair process, indels are introduced, achieving the goal of gene point mutations. Compared to ZFN, TALEN exhibits higher specificity and can efficiently achieve genome editing and modification. Additionally, the DNA binding of TALEN is modular, allowing convenient combination and modification to adapt to different target DNA sequences, making it flexible for application in various organisms. However, the modular design poses a challenge and requires certain professional knowledge and skills. Furthermore, TALEN possesses a certain degree of cytotoxicity, which can have adverse effects on cell growth and division. The emergence of the "mythical nuclease" TALEN quietly opened the door to the era of genetic programming.
In November 2022, the gene-editing company Cellectis announced preclinical data on its CAR-T cells based on TALEN gene editing. The US FDA has approved the IND application for the investigational product UCART20x22 to initiate a Phase 1/2a clinical trial, NatHaLi-01, for the treatment of recurrent or refractory non-Hodgkin's lymphoma (r/r NHL). UCART20x22 utilizes TALEN technology to knock out the TRAC gene and CD52 gene, reducing the risk of graft-versus-host disease and allowing the use of CD52 monoclonal antibodies in the patient's pretreatment protocol to enhance the engraftment, expansion, and durability of CAR-T cells.
3.The Dawn of the Age of Genomic Programming - CRISPR-Cas
In 1987, Japanese scientist Ishino Yoshizumi serendipitously discovered a series of seemingly disordered sequences sandwiched between identical sequences in his research on Escherichia coli. In 1993, Spanish scientist Francisco Mojica observed this repetitive structure in 20 different microorganisms. Over the years, the research on these sequences intensified, culminating in the Nobel Prize in Chemistry awarded to Doudna and Charpentier in 2020 for their groundbreaking work. These mysterious repeating sequences are known as Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR).
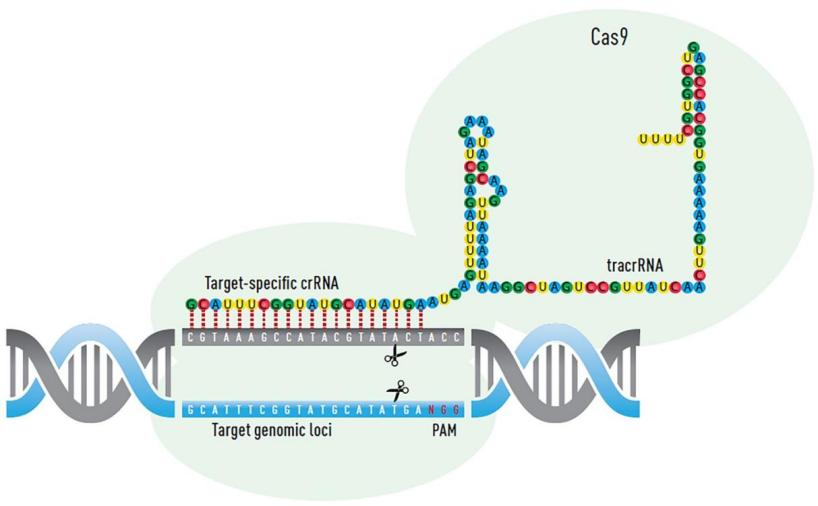
The CRISPR system is a nucleic acid enzyme system mediated by single-guide RNA. It specifically pairs CRISPR sequences with target sequences to guide the Cas9 protein to specific cleavage sites for cutting. Cellular repair mechanisms such as HDR or NHEJ are then employed for repair. HDR offers high precision but low efficiency and limited application, while NHEJ is the primary repair mechanism in cells. NHEJ repair may result in small insertions, deletions, and substitutions, enabling gene editing.
CRISPR technology boasts remarkable precision in identifying and cleaving target DNA sequences. Its high specificity, coupled with the ability to edit genes in a large number of cells in a short time frame, renders it applicable to a diverse range of organisms. Compared to other editing techniques, CRISPR offers significant advantages in terms of simplicity, efficiency, and cost. However, despite its high specificity, CRISPR technology is susceptible to off-target effects, leading to large deletions, translocations, and ruptures in genomic DNA. Additionally, when used in vivo, the Cas9 protein may trigger immune responses, compromising gene editing efficiency.
In May 2024, the Massachusetts Eye and Ear Infirmary and the Oregon Health & Science University jointly launched the EDIT-101 experimental gene editing therapy. This clinical trial involved 14 participants, with low-dose (2 participants), medium-dose (5 participants), and high-dose (5 participants) injections administered to 12 adults aged 17 to 63, and medium-dose injections given to 2 children aged 9 and 14. All participants suffered from LCA10 caused by mutations in the CEP290 gene. During the experiment, one eye of each participant received a single injection of EDIT-101, with no serious adverse events related to the treatment or procedure reported. There were also no dose-limiting toxicities observed. Using FST assessments, significant improvements in cone photoreceptor cells were observed in 6 participants, with 5 showing improvement in at least one other key secondary outcome. Nine participants (64%) demonstrated significant improvements in best-corrected visual acuity, red light sensitivity measured by FST, or activity performance test scores. Additionally, 6 participants reported significant improvements in vision-related quality of life scores.
4. Ushering in the Era of Precision Editing--Base Editor
In 2016, the laboratory of David R. Liu published a paper in Nature titled "Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage." This seminal work introduced the Base editor, a CRISPR/Cas9-based tool for single-base editing that enhances both the specificity and efficiency of the CRISPR system.
The Base editor system employs a fusion protein consisting of the nuclease-deactivated Cas9 (dCas9) and a cytidine deaminase. The cytidine deaminase targets the R-loop region formed by genomic DNA, where it binds to the single-stranded DNA (ssDNA) and deaminates cytosines (C) within a specific range to uracils (U). Subsequently, through DNA replication or repair mechanisms, these uracils are converted into thymines (T), effectively replacing CG base pairs with TA base pairs. This technique, known as CBE (Cytosine Base Editor), allows for the direct conversion of specific cytosine bases to thymine bases in the genome. In this study, the Liu team optimized the type of deaminase, its fusion location, and the linker sequence on the CRISPR-Cas9 backbone, leading to the development of the first-generation tool, rAPOBEC1-XTEN-Cas9, also known as BE1. To further enhance editing efficiency and reduce the frequency of insertions and deletions (indels), the team incorporated UGI (Uracil DNA glycosylase inhibitor) at the C-terminus of Cas9, resulting in the second-generation tool, rAPOBEC1-XTEN-Cas9-UGI, or BE2. Aiming to surpass the theoretical limit of 50% editing efficiency, the team substituted dCas9 with nCas9 (nickase Cas9), which possesses single-strand cleavage activity, culminating in the third-generation tool, rAPOBEC1-XTEN-nCas9-UGI, or BE3.
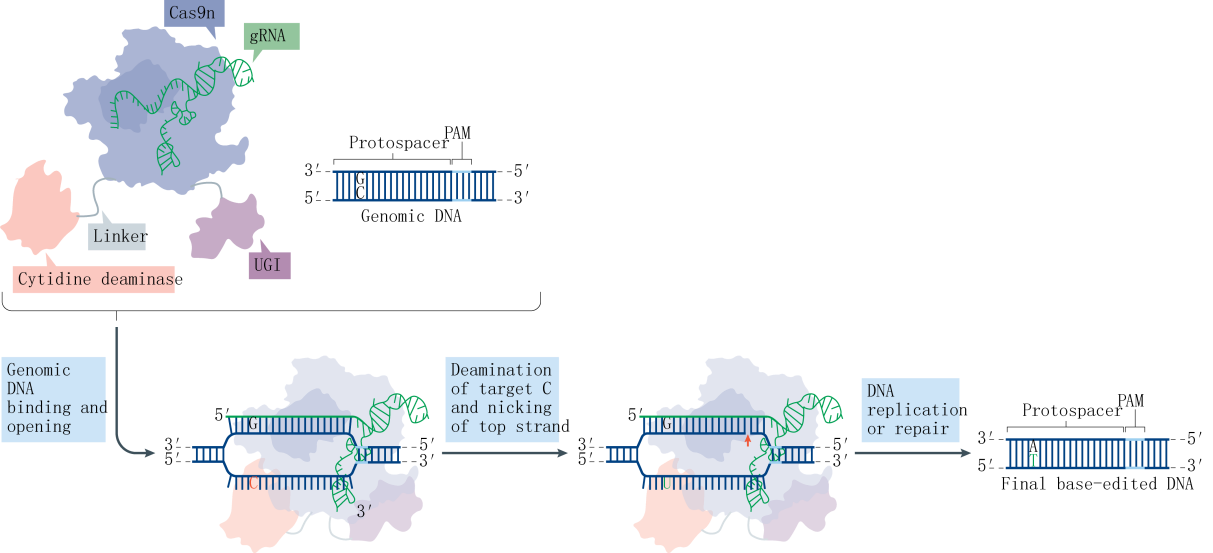
Research indicates that a majority of single-base genetic diseases are caused by G-A mutations. Following the establishment of the CBE system, the subsequent challenge was the development of an Adenine Base Editor (ABE) system. A significant hurdle in developing the ABE was the absence of naturally occurring DNA adenosine deaminases. To overcome this obstacle, the David Liu team selected the TadA protein from Escherichia coli as a substrate and designed an antibiotic screening system for directed evolution. After seven rounds of evolution, the team successfully isolated an artificially evolved adenosine deaminase. When the nCas9 fused with the adenosine deaminase is guided by sgRNA to target genomic DNA, the adenosine deaminase binds to the ssDNA and deaminates adenines (A) within a specific range to inosines (I). Inosines are recognized and replicated as guanosines (G) at the DNA level, ultimately facilitating the direct replacement of AT base pairs with GC base pairs.
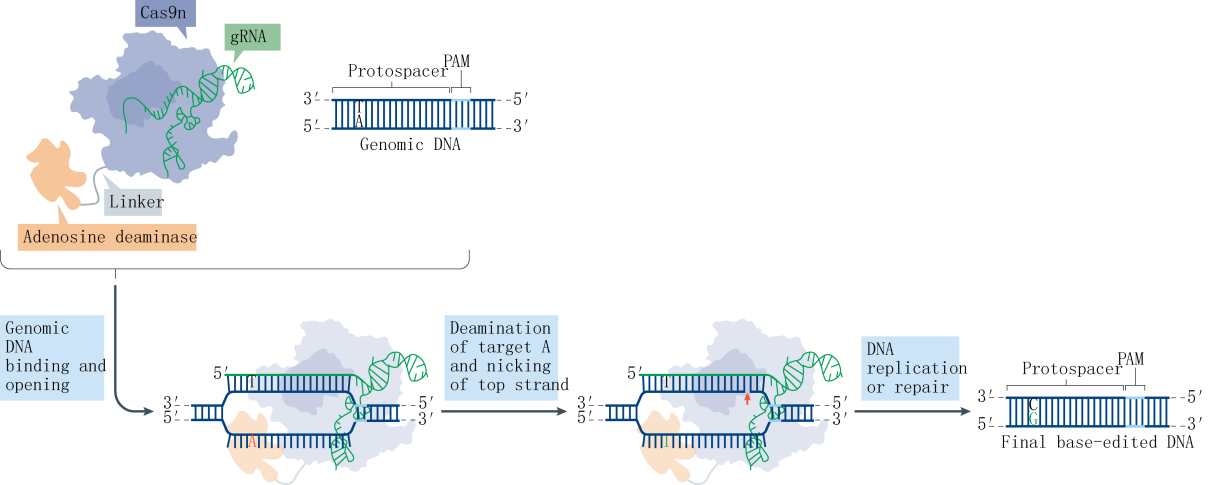
Further statistical analysis revealed that the probability of indels caused by CBE is significantly higher than that of ABE. Upon investigation, it was discovered that cells contain UNG (uracil N-glycosylase). When CBE deaminates cytosine (C) to uracil (U), UNG recognizes and removes the uracil from the genome, creating an a basic site that leads to the formation of indels. To address this issue, the David Liu team optimized the BE3 linker and increased the number of UGI fusions, resulting in the fourth-generation tool, rAPOBEC1-nCas9-UGI-UGI, or BE4. Subsequently, further codon optimization and the addition of NLS (Nuclear Localization Sequence) were performed on BE4 to enhance its efficiency further, yielding BE4max. Recently, another team proposed an alternative approach and developed the GBE system. GBE incorporates exogenous UGI (uracil glycosylase inhibitor) into the CBE framework to suppress the activity of UNG, protecting the uracil base and improving the efficiency of conversion from cytosine to uracil and, ultimately, to thymine.
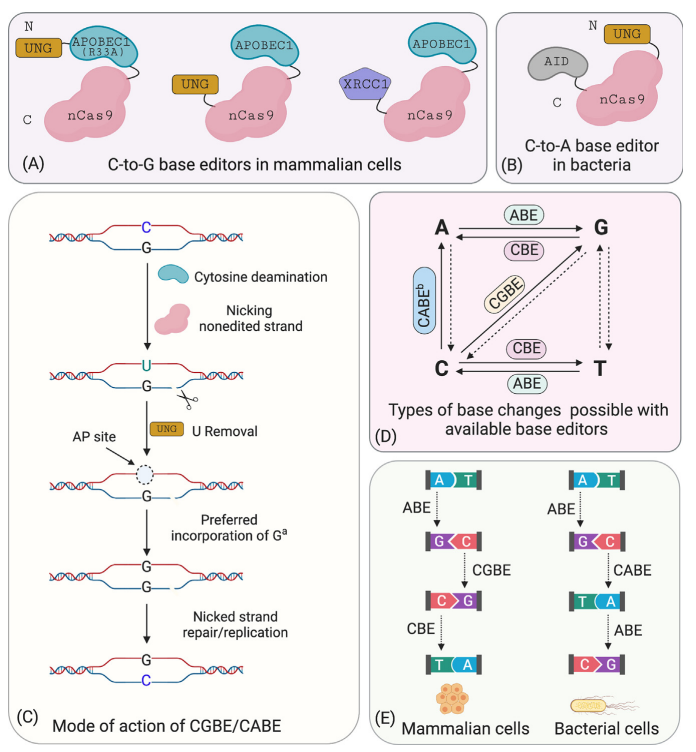
The combined use of CBE and ABE can effectively perform four types of base conversions (C → T, G → A, A → G, T → C). However, beyond these four base conversions, it is not possible to achieve the other eight base conversions (C → A, C → G, G → C, G → T, A → C, A → T, T → A, T → G) as well as base insertions and deletions. Additionally, due to the specific range of action for the respective fusion proteins, similar bases near the target base may undergo base conversion, thereby reducing the specificity of editing efficiency.
In January 2024, a clinical trial (IIT) conducted by researchers at the First Affiliated Hospital of Guangxi Medical University on the base editing drug CS-101 for severe β-thalassemia successfully cured the first patient. The treatment led to sustained independence from blood transfusions for over two months. Eight weeks post-treatment, the fetal hemoglobin concentration of the patient rose to ∼95 g/L, with its proportion increasing to ∼81% and the proportion of F cells expressing fetal hemoglobin rising to ∼80%. CS-101 involves collecting autologous hematopoietic stem cells from patients and using the highly precise transformer Base Editor (tBE) to precisely edit the HBG1/2 promoter region in the patient's autologous hematopoietic stem cells. This process mimics naturally occurring beneficial base mutations found in healthy populations, reactivating the expression of γ-globin, rebuilding the oxygen-carrying function of hemoglobin, and then reinfusing the edited hematopoietic stem cells back into the patient's body. This procedure enables the patient's own hemoglobin concentration to reach healthy levels, thus completely eliminating the need for blood transfusions.
5. Milestone in Precision Editing - Prime Editing
In 2019, the David Liu team published a paper in the journal Nature titled "Search-and-replace genome editing without double-strand breaks or donor DNA[2]," marking the advent of the Prime Editing tool.
The Prime Editor complex recognizes PAM under the guidance of pegRNA and binds to the target DNA strand complementary to the guide RNA. The Cas9 enzyme creates a nick between the third and fourth nucleotides upstream of the original PAM sequence; the cleaved single-stranded DNA binds to PBS, and the reverse transcriptase becomes active, conducting reverse transcription along the PBS to generate the edited sequence. The edited sequence competes with the original target DNA sequence to bind to the other strand through base pairing; unbound sequences will be displaced and eventually excised. This competitive binding results in two outcomes: either the original target DNA sequence remains while the edited sequence is excised, in which case the target DNA is not successfully edited and may be recognized again by the complex, or the edited sequence remains while the original target DNA sequence is excised. In this latter case, since the edited sequence does not match the sequence of the other strand, a mismatch is formed at the editing site, prompting the cellular DNA mismatch repair mechanism to intervene. The repair outcomes are also twofold: either the editing site is modified, reverting to the initial state of the target DNA, or the other strand is repaired, producing an edited double-stranded DNA, successfully achieving the goal of precise gene editing.
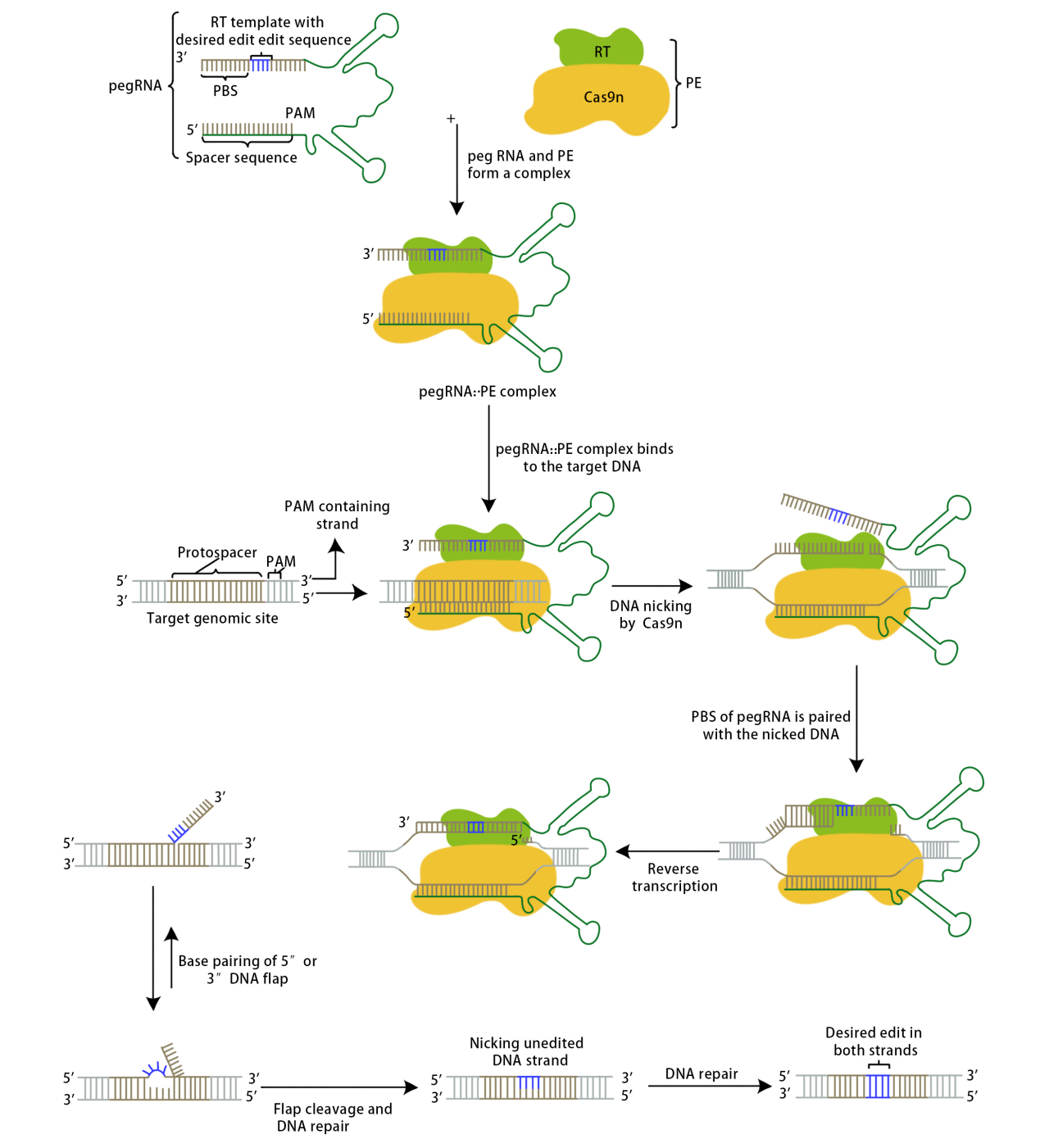
David Liu's team conducted experiments related to PE1, PE2, and PE3. The PE1 system used a wild-type M-MLV reverse transcriptase fused to either end of the Cas9 H840A nickase via a flexible linker, co-transfected with a pegRNA into HEK293T cells. Initial attempts did not detect editability; later, by extending the primer binding site (PBS) in the pegRNA to 8-15 bases, PE1 was able to detect convertible point mutations at the HEK3 target site. To improve the efficiency of DNA synthesis, researchers engineered the RT in PE1, constructing 19 PE1 variants and evaluating their editing efficiency in human cells. Introducing specific M-MLV RT mutations (D200N+L603W+T330P, known as M3) increased the average conversion and insertion editing efficiency by 6.8 times across five genomic sites compared to PE1. Further addition of T306K and W313F mutations formed PE2, which improved the editing efficiency by 1.3 to 3.0 times over PE1 for six conversion or insertion edits across five genomic sites. The PE3 system enhanced editing efficiency by using the Cas9 H840A nickase to make nicks on the non-edited strand; testing it on five genomic sites showed an improvement of 1.5 to 4.2 times in editing efficiency compared to PE2, reaching up to 55%. Further experiments revealed that nickases often create nicks on both strands of DNA, triggering the double-strand repair mechanism and introducing indels. Redesigning the newly added sgRNA ensured that nicks were made on the non-edited strand only after editing the PAM strand, reducing the formation of indels by an average of 13 times (0.74%) without significantly decreasing editing efficiency in the PE3b system.
In 2021, David Liu collaborated with the Adamson team from Princeton to develop PE4, PE5, and PEmax systems, publishing an article titled: "Enhanced prime editing systems by manipulating cellular determinants of editing outcomes [3]." They used CRISPRi screening to discover that the DNA mismatch repair (MMR) pathway inhibited the efficiency and accuracy of Prime editing. Knocking down components of the MMR complex (such as MSH2, MSH6, MLH1, and PMS2) was observed to enhance Prime editing efficiency. PE4 and PE5 were developed on the basis of PE2 and PE3 by co-expressing an engineered MMR inhibitory protein (MLH1dn) that lost its endonuclease function, transiently inhibiting the activity of mismatch repair enzymes. In six MMR-normal cell types, including induced pluripotent stem cells (iPSCs) and primary T cells, the PE4 and PE5 systems increased editing efficiency by an average of 7.7 and 2.0 times compared to PE2 and PE3 systems, respectively, while also increasing the edit/indel ratio by 3.4 times. By optimizing the reverse transcriptase codon, adding some amino acid mutations to SpCas9 to enhance enzymatic cutting efficiency, adding NLS peptide segments at both ends of the entire enzyme to increase the adsorption force of the composite enzyme to nucleic acids, and adjusting the length and composition of the peptide chain link between nCas9 and the reverse transcriptase, the PEmax architecture was developed. This architecture improved editing efficiency by an average of 2.5 times in HeLa cells and 1.2 times in HEK293T cells. Using the PE4max and PE5max systems for editing on six genomic sites related to sickle cell anemia, prion disease, CDKL5 deficiency syndrome, HIV infection, and T cell transfer therapy, significant improvements in editing efficiency and reductions in indel byproducts were observed.
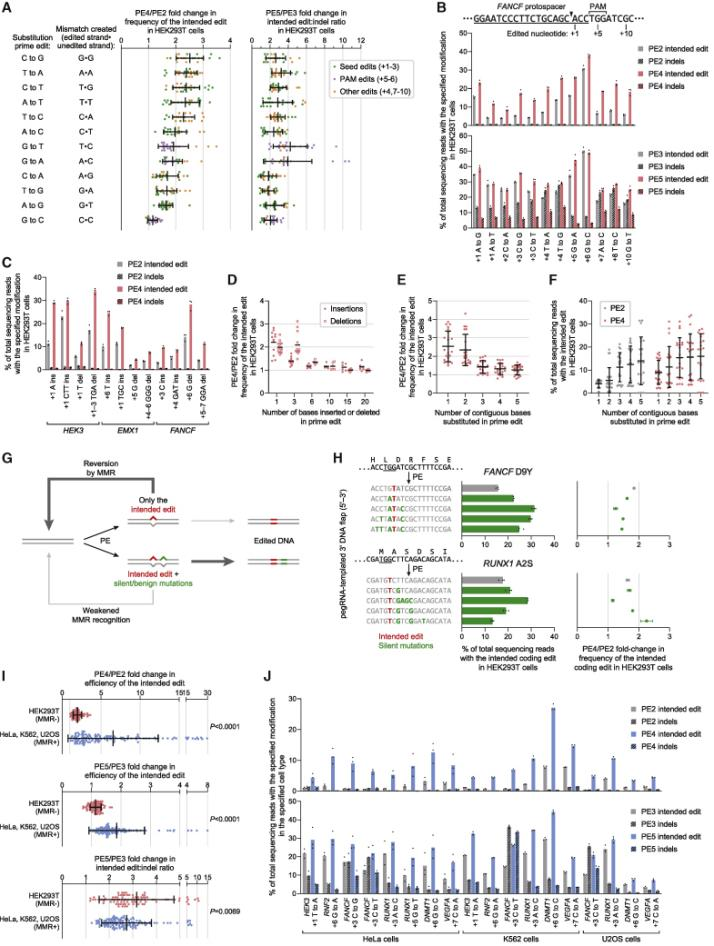
In August 2023, the David Liu team published an article in Cell titled: "Phage-assisted evolution and protein engineering yield compact, efficient prime editors [4]," where they developed a new PE6 and its variants PE6a-g. Using Phage-Assisted Continuous Evolution (PACE), phages carrying the original PE editor underwent thousands of generations of evolution over several hundred hours, rapidly producing reverse transcriptase with novel structures, resulting in the PE6 editor that is smaller and more efficient than the current PEmax editor. PE6 was tested in mice through an AAV delivery system and achieved up to 40% loxP sequence insertion in the mouse cerebral cortex, which is a 24-fold improvement over previous PE editors. Additionally, the David Liu team developed various PE6 variants (PE6a-g), each with its specific advantages, to meet different types of gene editing needs and provided a decision tree to help researchers choose the appropriate PE6 variant based on their specific editing requirements.
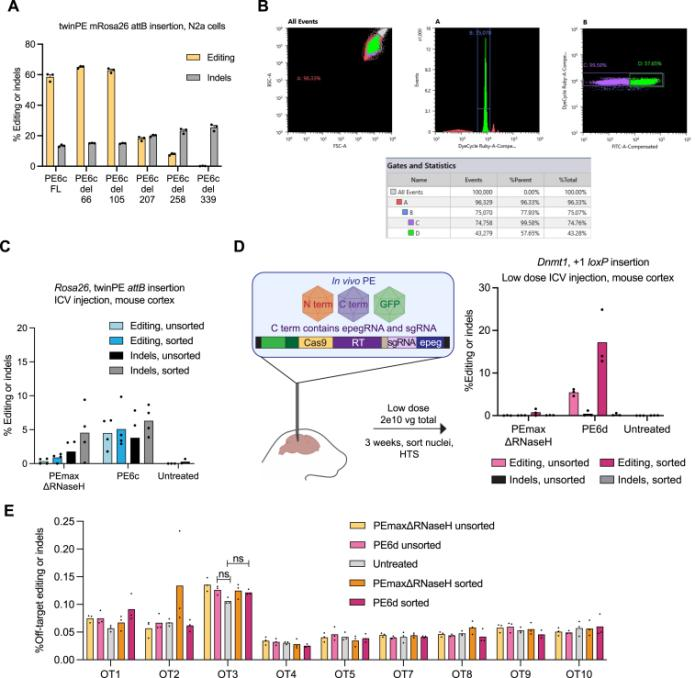
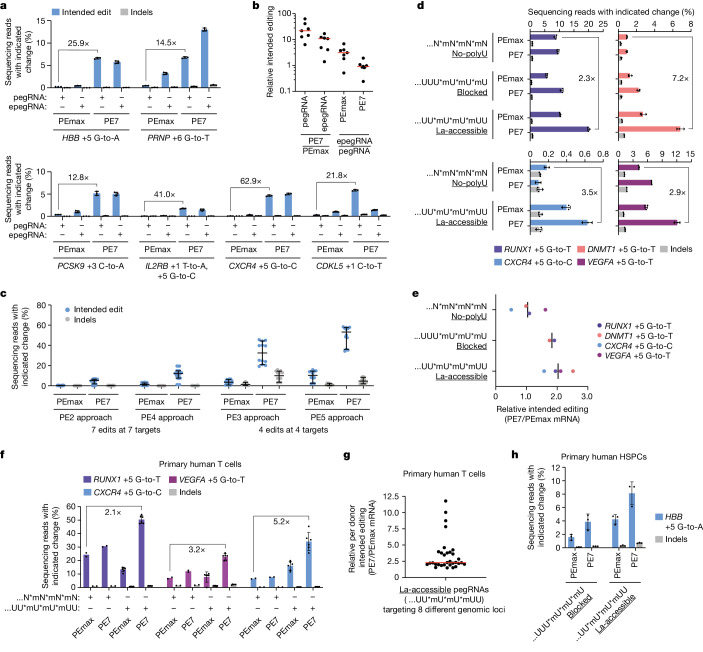
In April 2024, the team led by Britt Adamson and Lance R. Parsons from Princeton University in the United States published an article in Nature titled "Improving prime editing with an endogenous small RNA-binding protein [5]," where they discovered a key regulatory factor that affects PE efficiency: La protein, and developed a new PE system called PE7. PE7 demonstrated improved efficiency across eight different genomic loci (such as the HBB gene associated with sickle cell disease, the PRNP gene associated with prion diseases, and the PCSK9 gene associated with familial hypercholesterolemia), three cell lines (HEK293T, HeLa, and U2OS), and different types of editing (single-nucleotide substitutions, insertions, or deletions of 15 base pairs). Notably, the most significant improvements were observed in HeLa and U2OS cells with intact mismatch repair (MMR) systems. Compared to PEmax, PE7 showed a median improvement of 21.8-fold and 10.8-fold when used with pegRNA and epegRNA for editing in U2OS cells, respectively. When used for editing in primary human T cells and CD34+ hematopoietic stem and progenitor cells (HSPCs), PE7 paired with La-accessible pegRNAs demonstrated higher target editing efficiency compared to PEmax. The researchers also tested PE7 mutants with impaired RNA-binding ability of La (1 – 194) and found that these mutations abolished the improvement observed when La(1 – 194) was fused to PEmax, confirming the model that La protein promotes prime editing through interactions with the 3' end of pegRNA.
On April 29, 2024, the first Prime Editing gene-editing therapy was approved for clinical trials. Prime Medicine, a company led by David Liu, announced that its Investigational New Drug (IND) application for a prime editing therapy to treat Chronic Granulomatous Disease (CGD) had been approved by the U.S. Food and Drug Administration (FDA), marking the initiation of global phase 1/2 clinical trials. This phase 1/2 clinical trial will assess the safety, biological activity, and preliminary efficacy of the prime editor therapy in adult and pediatric patients with CGD. The initial clinical trial participants will be adults with stable conditions; once safety and biological activity are demonstrated in these patients, the trial will expand to include patients experiencing persistent infections or severe inflammation, as well as adolescents and children. Clinical trial participants will undergo safety follow-ups, including hematopoietic system engraftment and reconstitution, early biomarkers of immune function recovery, and long-term resolution and prevention of CGD-related infection and inflammation complications.
Conclusion
Reflecting on the leapfrog development of gene-editing technology from ZFN to Prime Editing, we have witnessed tremendous progress in precisely manipulating the genetic code of life. These technologies not only provide us with a window to deeply understand the mysteries of life but also bring unprecedented possibilities to disease treatment, crop improvement, and other fields. Although each technology has its own advantages and limitations, it is the continuous improvement and innovation of these technologies that drive the deep development of life science research. With the ongoing advancements in biotechnology and the integration of interdisciplinary studies, gene-editing technology will demonstrate its powerful application potential in more fields. We look forward to these technologies becoming more mature, efficient, and safe, making greater contributions to human health and well-being.
- EDITGENE has been deeply involved in the field of gene editing for many years, specializing in the underlying research and development of CRISPR/Cas. Based on the Prime Editing technology, we have upgraded and developed the Bingo ™ precision editing gene site-directed mutagenesis platform, providing precise and efficient customized services for gene site-directed mutagenesis cells to various scientific research institutions and enterprises worldwide.

Recent Edi-blogs:
2. 【 New Trends in Gene Editing 】 CTLA4, MUC1, and mTOR Knockout Cells
3. [Mother’s Day bundle Day Bundle | EDITGENE Cell Line Bundle]
Follow us on social media
![]()
![]()
![]()
![]()
![]()
Contact us
+ 833-226-3234 (USA Toll-free)
+1-224-345-1927 (USA)
+86-13250544625(Intl)info@editxor.com
References :
[1] Komor A C, Kim Y B, Packer M S, et al. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage[J]. Nature, 2016, 533(7603): 420-4.
[2] Anzalone A V, Randolph P B, Davis J R, et al. Search-and-replace genome editing without double-strand breaks or donor DNA[J]. Nature, 2019, 576(7785): 149-57.
[3] Chen P J, Hussmann J A, Yan J, et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes[J]. Cell, 2021, 184(22): 5635-52.e29.
[4] Doman J L, Pandey S, Neugebauer M E, et al. Phage-assisted evolution and protein engineering yield compact, efficient prime editors[J]. Cell, 2023, 186(18): 3983-4002.e26.
[5] Yan J, Oyler-Castrillo P, Ravisankar P, et al. Improving prime editing with an endogenous small RNA-binding protein[J]. Nature, 2024, 628(8008): 639-47.
Tag
Share






Comment (4)