[Literature Review] CRISPR KO Screening Reveals Key Genes Responsible for Chemotherapy Resistance
CRISPR knockout screening

In June this year, a research team led by Professor Fei Teng from the College of Science and Health at Northeastern University, in collaboration with Professor Jun Huang from the Department of Colorectal Surgery at the Sixth Affiliated Hospital of Sun Yat-sen University and Professor Huajun Wu from the Peking University Institute of Clinical Medicine/Beijing Cancer Hospital, published a research paper titled "CRISPR screens reveal convergent targeting strategies against evolutionarily distinct chemoresistance in cancer" in Nature Communications .
This study utilized 30 whole-genome CRISPR knockout screens and 26 druggable gene library screens to systematically identify chemotherapy resistance-related genes in various tumor cells for seven common chemotherapy drugs. The research successfully unveiled the molecular mechanisms behind cancer cells' resistance to multiple chemotherapy drugs and provided new strategies for future cancer treatments.

Original Article Link: https://doi.org/10.1038/s41467-024-49673-4
I. Genome-Wide CRISPR Screening for Chemotherapy Resistance Genes
To systematically identify loss-of-function genes that drive chemotherapy resistance in cancer, the researchers employed CRISPR knockout screening technology. They evaluated the effects of seven commonly used chemotherapy drugs in colorectal, breast, and lung cancer cells. By using lentiviral vectors to transduce Cas9 nuclease and sgRNA into the cells, the sgRNAs targeted 18,436 protein-coding genes in the human genome.
The results showed that in screens treated with the vehicle control (DMSO) without selective pressure, core essential genes exhibited strong negative selection. Each drug applied a distinct selective pressure, which was assessed by the Gini index and the uniformity of sgRNA count distribution.
The impact of gene knockouts was quantified using MAGeCK to calculate RRA scores, where lower negative values indicated stronger negative selection and higher positive values indicated stronger positive selection.
"Chemotherapy resistance genes" were defined as genes whose loss of function induced resistance to chemotherapy drugs in the screened cells. These genes could be identified from positive selection hits, and their function might inherently suppress or inhibit chemotherapy resistance.
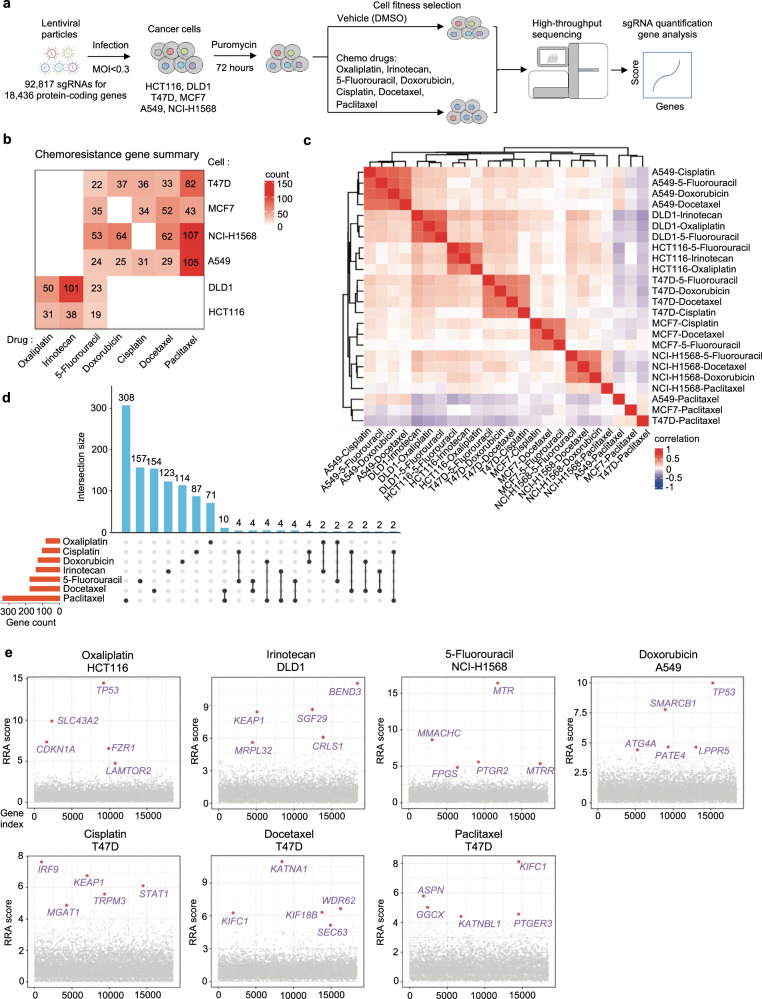
Figure 1: Genome-Scale Identification of Chemoresistance Genes
II. A Systematic View of the Genetic Landscape of Chemoresistance
Through CRISPR knockout screens across multiple cancer cell lines, researchers identified dozens of genes associated with resistance to various chemotherapy drugs. Analysis of the results revealed that these chemoresistance genes primarily clustered based on cell origin rather than drug type, highlighting the importance of genetic background in determining chemotherapy resistance.
Although there was some overlap in chemoresistance genes across different cell lines, most resistance genes exhibited cell-specific differences.By integrating results across cell lines, the researchers generated drug-specific resistance gene lists, uncovering how cellular genetic background and drug mechanisms influence the variation in resistance genes.
Further functional enrichment and network analysis revealed that functions such as cell cycle regulation, DNA damage response, and fibroblast proliferation played roles in multidrug resistance, while specific mitochondrial-related mechanisms were closely linked to irinotecan resistance.
These data systematically provide insights into the mechanisms by which chemoresistance develops at the gene or pathway level.
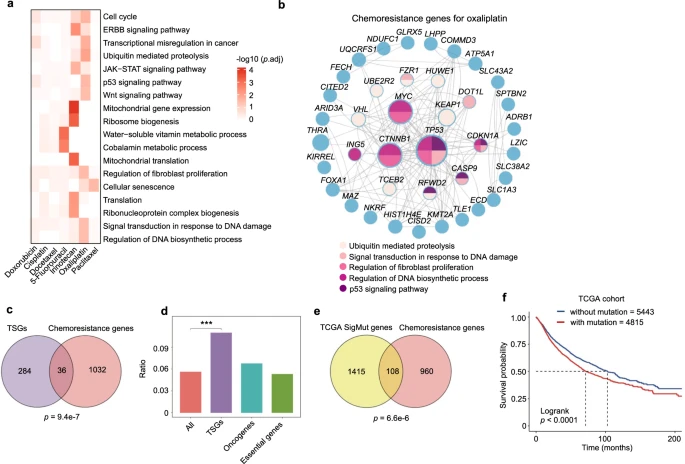
Figure 2: A Systematic View on Chemoresistance Genes.
III. Clinical Relevance of Chemotherapy Resistance Genes
To explore the relationship between chemoresistance genes identified in vitro and human clinical data, the researchers first analyzed the association between chemoresistance genes and tumor suppressor genes (TSGs), as the loss of TSG function is often linked to cancer development.
Using the TSG list from the COSMIC Cancer Gene Census, they found a significant overlap between chemoresistance genes and TSGs, with no apparent association with oncogenes or pan-essential genes.
Although previous studies have reported connections between TSGs and resistance, such as TP53, this study systematically confirmed the importance of TSGs in chemoresistance.By analyzing somatic mutation data from The Cancer Genome Atlas (TCGA), the researchers discovered that many chemoresistance genes are frequently mutated in tumors, and these mutations are associated with poorer patient survival.
Frequently mutated resistance genes included known drug response regulators (such as TP53 and MED12) and other less recognized genes. Mutations or low expression of certain genes were correlated with lower survival rates in specific cancer types, further supporting the clinical relevance of the findings.
Additionally, in colorectal cancer cells treated with oxaliplatin, the expression levels of key resistance genes such as KEAP1, SLC43A2, or TP53 were significantly downregulated in recurrent tumors.
Overall, the mutational characteristics and/or expression status of chemoresistance genes may serve as potential biomarkers for predicting chemotherapy response.
IV. Rapid Derivation of Chemoresistant Cells through Evolutionarily Distinct Pathways
Traditional approaches to studying chemoresistance typically involve directly comparing resistant and sensitive cells with different genetic backgrounds or deriving matched resistant cells from sensitive parental cells through prolonged drug exposure.
In this study, the researchers rapidly and efficiently established multiple chemoresistant cell lines by directly knocking out specific chemoresistance genes.The results indicated that the sensitivity gene profiles varied across different drugs, and for certain drugs like irinotecan, sensitizing genes were not identified due to the screening pressure.
There was more overlap in sensitizing genes than in resistance genes across different drugs, with key sensitizing genes such as NEK7, KDM5C, KLF5, and BCL2L1 repeatedly appearing across different drugs, indicating a common vulnerability in multidrug resistance. Enrichment analysis suggested that targeting certain pathways could enhance therapeutic efficacy.
Although sensitizing genes differed between cell lines, the study revealed the diverse genetic mechanisms underlying resistance, highlighting the need to explore multiple chemoresistance pathways simultaneously to discover effective treatment strategies.
Additionally, the chemoresistant cells established through gene knockout demonstrated that the loss of a single gene could quickly lead to resistance without the need for drug adaptation. This not only validated the drug response phenotypes of top chemoresistance hits but also provided a valuable resource for the rapid generation of various chemoresistant cell lines.
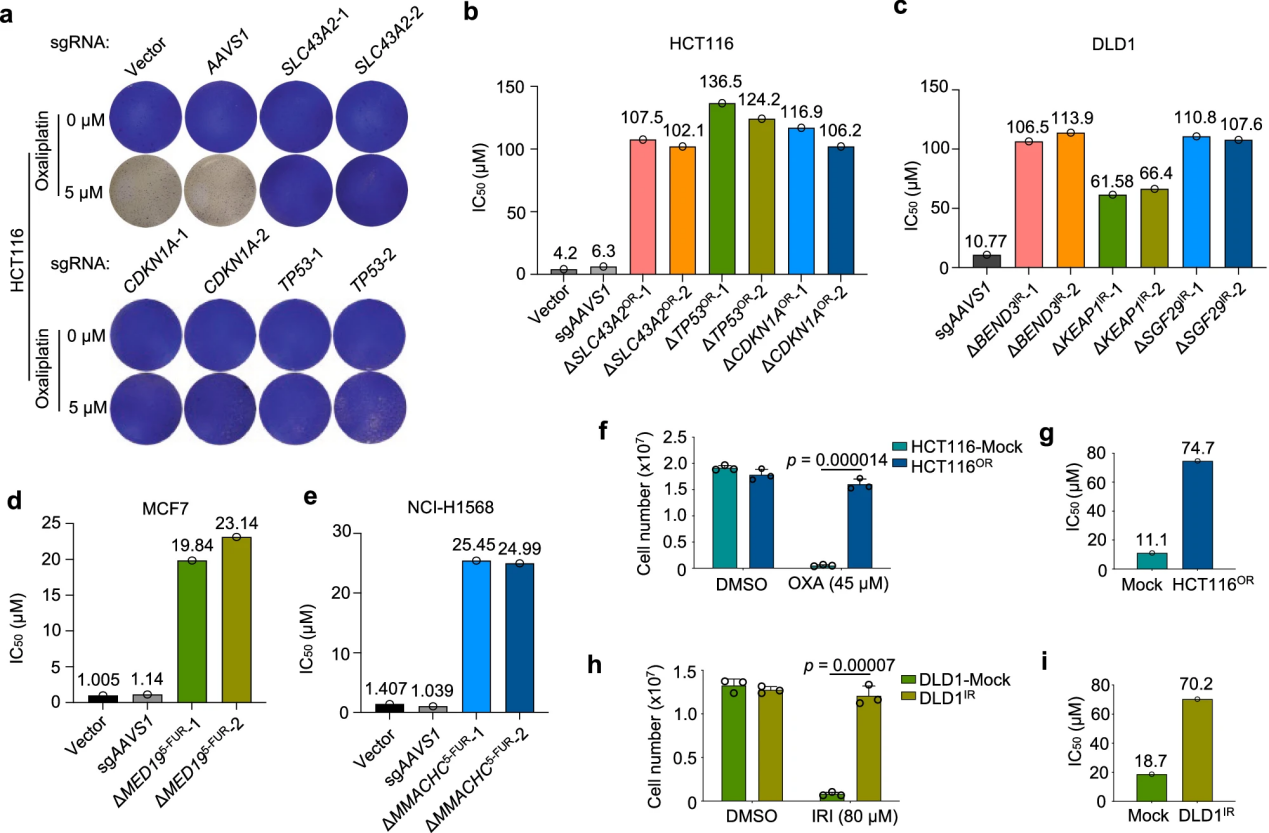
Figure 3: Rapid Derivation of Chemoresistant Cells
V. Cellular Characteristics of Chemoresistant Cells
Researchers investigated the molecular mechanisms of chemoresistance using resistant cell lines established either by knocking out specific genes or through long-term drug selection. Using oxaliplatin resistance as a model, the study found that resistant cells from different sources did not exhibit significant morphological changes or noticeable decreases in survival rates.
However, cell cycle analysis revealed that oxaliplatin significantly reduced the proportion of S-phase cells and caused cell cycle arrest at different doses. Resistant cells developed resistance by disrupting the p53/p21 signaling pathway to counteract these effects.
Additionally, while ERK signaling was suppressed in resistant cells, their apoptosis rates did not change significantly. Similarly, irinotecan-resistant cells exhibited comparable resistance characteristics, but no significant changes were observed in ERK signaling or other pathways. This suggests that targeting cell growth-related kinases may not be an effective strategy for overcoming irinotecan resistance in colorectal cancer.
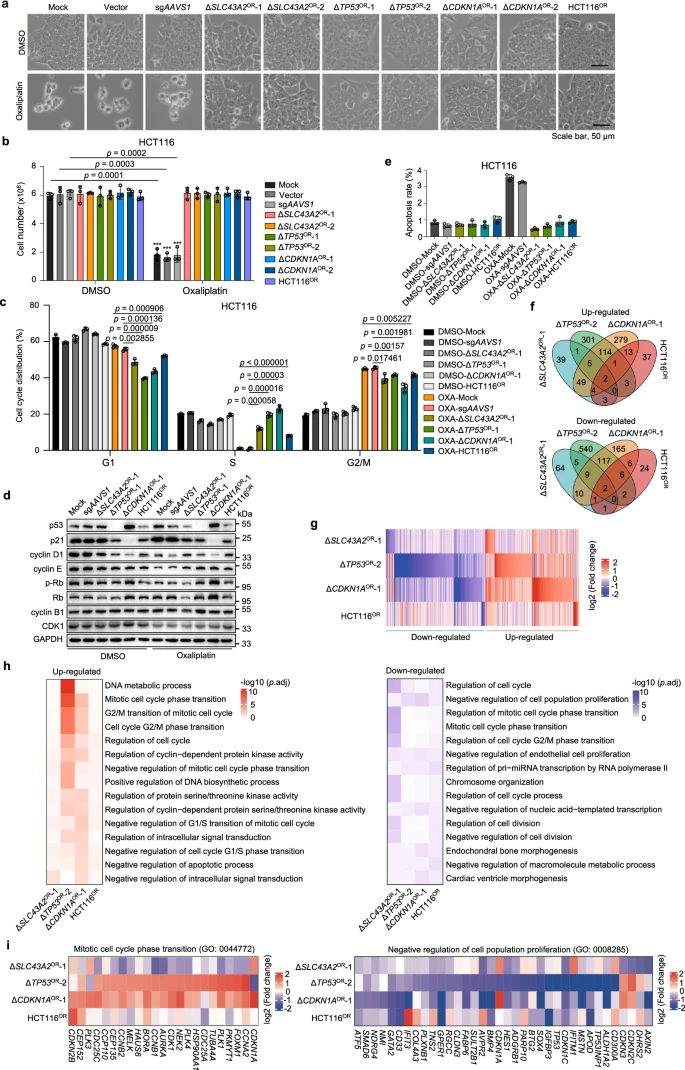
Figure 4: Cellular and Molecular Features of Oxaliplatin-Resistant Cells
VI. Gene Expression Characteristics of Oxaliplatin or Irinotecan Resistance
To characterize the intrinsic molecular features of chemoresistance, the researchers conducted transcriptome analysis through RNA sequencing on multiple chemoresistant cell lines, comparing the differential gene expression profiles between resistant models and sensitive cells.
The results revealed significant differences in gene expression patterns among various oxaliplatin-resistant cells, with some cells sharing more differentially regulated genes, particularly those under the regulation of p53 and p21.
Although gene expression profiles varied among resistant cells, cell cycle-related genes were consistently upregulated across multiple resistant cell lines, indicating that driving cell cycle progression is a primary mechanism for resisting oxaliplatin toxicity.
For irinotecan-resistant cells, significant overlap in gene sets was observed between different cells, with genes related to cytoskeletal organization, cell cycle, and DNA damage response being upregulated in resistant cells. This suggests that these pathways play a crucial role in chemoresistance.
Further analysis indicated that shared upregulated genes might be associated with lower survival rates, while positively selected genes in resistant cells tended to be downregulated, reinforcing the functional loss effects of these genes in driving chemoresistance.
VII. Second-Round CRISPR Screening for Druggable Targets
To identify potential druggable targets, the researchers constructed a new CRISPR knockout library containing 12,000 sgRNAs targeting 1,716 druggable genes in the human genome. This library was used to screen four oxaliplatin-resistant cell lines based on HCT116 and two resistant cell lines with different backgrounds (DLD1 and HCT8), along with their sensitive control cells.
Data analysis revealed that druggable genes with stronger negative selection in resistant cells, particularly the cell cycle regulator PLK4, had a significant impact on oxaliplatin resistance. Similarly, screening for irinotecan resistance found that genes related to mitochondria and cellular respiration were enriched in resistant cells.
These results highlighted common vulnerabilities in different chemoresistance mechanisms, and the druggable targets or pathways identified through this approach offer a preliminary and practical option for using single-agent strategies to overcome chemoresistance.
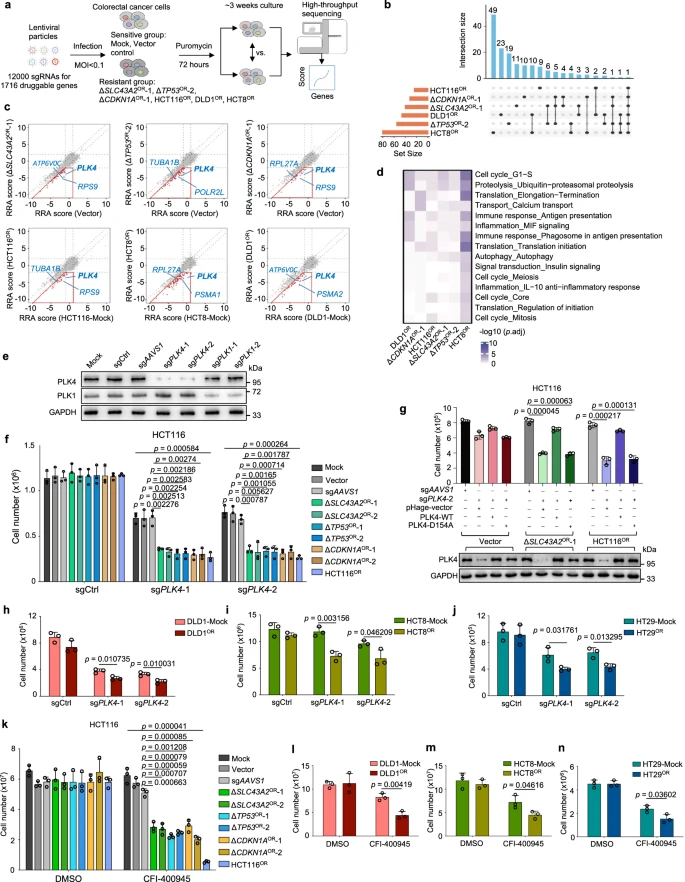
Figure 5: Interrogation of Druggable Targets Against Oxaliplatin Resistance by Second-Round CRISPR Screens
VIII. Selective Dependency of Oxaliplatin-Resistant Cells on PLK4
To validate the effectiveness of targeted strategies, the researchers focused on PLK4, a gene that emerged as a potential target in colorectal cancer and oxaliplatin resistance. PLK4 encodes a serine/threonine protein kinase that plays a crucial role in cell cycle regulation, centriole duplication, and cytokinesis, and is associated with resistance to various chemotherapy drugs.
To explore how PLK4 functions in oxaliplatin-resistant cells, the researchers knocked out PLK4 or its homolog PLK1. They found that PLK4 knockout significantly reduced the growth of seven oxaliplatin-resistant cell lines, whereas PLK1 had a lesser impact. Further rescue experiments demonstrated that the kinase activity of PLK4 was essential for this growth inhibition phenotype. To identify downstream PLK4 target genes, the researchers conducted RNA sequencing after CRISPR-mediated PLK4 knockout. They discovered a varying number of target genes, but no significant differences between the resistant and sensitive groups.
Functional enrichment analysis revealed that RNA processing-related genes were enriched among PLK4 upregulated targets, while specific enrichments in resistant cells were related to fundamental cellular functions.
The researchers hypothesized that PLK4-regulated pericentriolar material might be more critical than centriole biogenesis in distinguishing the spindle collapse phenotype between oxaliplatin-resistant and sensitive cells.
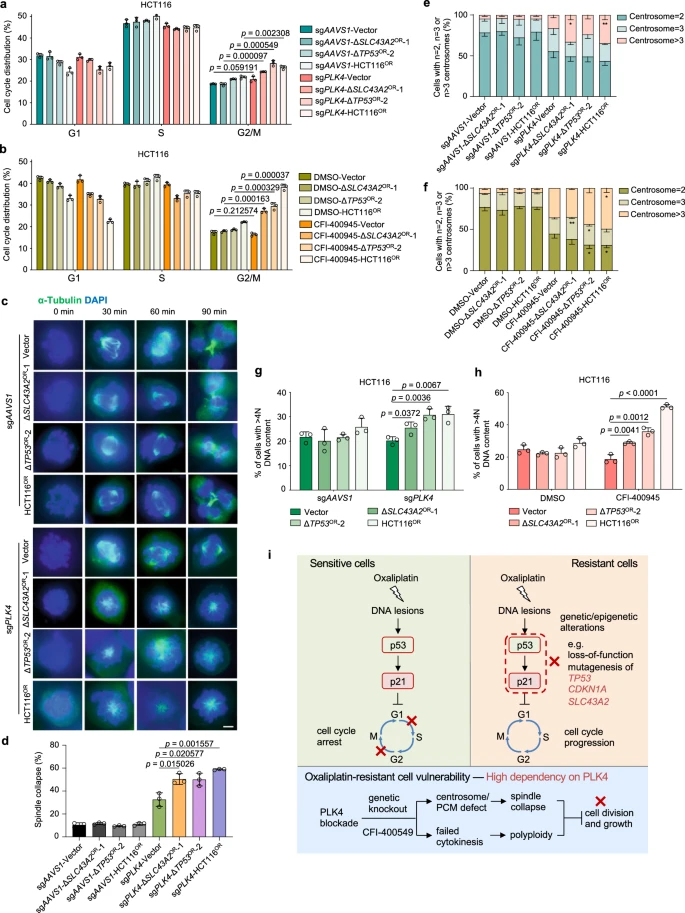
Figure 6: Selective Dependency on PLK4 for Oxaliplatin-Resistant Cells
IX. Mechanistic Insights into Oxaliplatin Resistance and PLK4 Dependency
Through a combination of CRISPR screening and cellular and molecular characterization of various oxaliplatin-resistant models, researchers have begun to unravel the mechanisms underlying oxaliplatin resistance in colorectal cancer.
In sensitive cells, oxaliplatin induces DNA damage by forming DNA adducts, which activates the p53-p21 pathway, leading to cell cycle arrest in the G1 and G2/M phases, thereby halting cell growth. However, when genetic or epigenetic alterations occur, such as the loss of TP53 or CDKN1A, cell cycle regulation becomes disrupted, resulting in resistance.
While mechanisms such as drug transport, detoxification, DNA repair, and cell death also contribute to resistance, the study indicates that cell cycle regulation is a central mechanism of oxaliplatin resistance in colorectal cancer. In resistant cells, PLK4 functions as a critical vulnerability because these cells exhibit heightened dependency on PLK4. The loss or inhibition of PLK4 leads to spindle assembly failure and issues with cell division.
Targeting PLK4 could therefore provide a therapeutic opportunity for treating oxaliplatin-resistant colorectal cancer.
X. Targeting PLK4 in Xenograft Models and Clinical Samples
To further explore the therapeutic potential of targeting PLK4 to counteract oxaliplatin resistance, researchers conducted experiments using both oxaliplatin-sensitive and -resistant mouse models, monitoring tumor growth. The results demonstrated that CFI-400945, a PLK4 inhibitor, significantly suppressed the growth of oxaliplatin-resistant tumors more effectively than it did in sensitive tumors.
PLK4 is frequently mutated across various cancers, including colorectal cancer, and its RNA expression is commonly elevated in colorectal tumors. By establishing seven patient-derived organoid (PDO) models, the researchers found that PLK4 protein levels were higher in these models compared to normal tissues. Furthermore, the oxaliplatin-resistant PDOs showed greater sensitivity to CFI-400945.
These findings suggest that CFI-400945 as a monotherapy could be a promising strategy to overcome oxaliplatin-induced chemotherapy resistance in colorectal cancer.
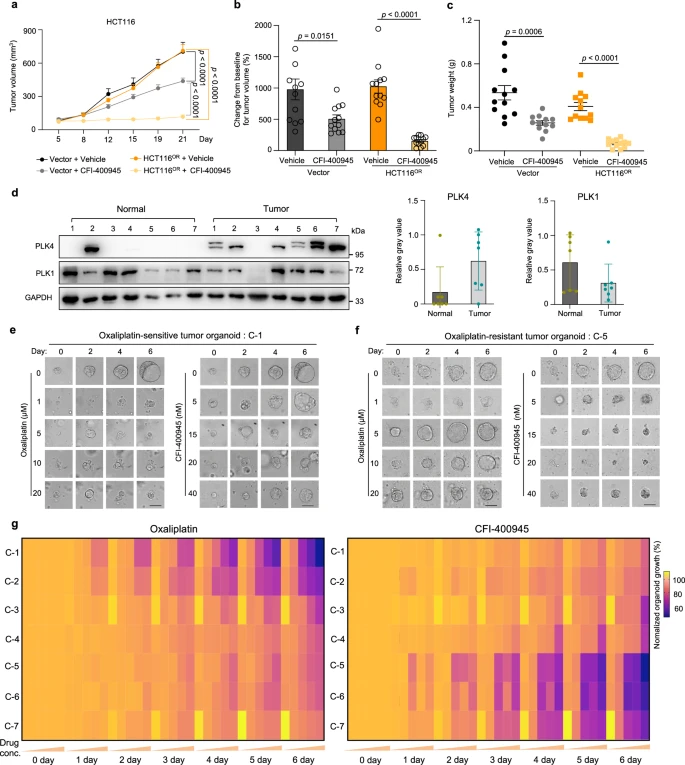
Figure 7: Validation of Efficacy of the PLK4 Inhibitor CFI-400945 Against Oxaliplatin Resistance in Mice and at Clinical Levels
In summary, researchers utilized CRISPR KO screening technology to uncover the genetic causes and mechanisms of chemotherapy drug resistance, highlighting key drivers such as cell cycle abnormalities, DNA damage response, and tumor microenvironment remodeling. They also identified promising biomarkers and therapeutic strategies targeting these resistance mechanisms. These findings provide valuable insights into understanding and overcoming chemotherapy resistance, with a particular focus on validating PLK4 as a therapeutic target for oxaliplatin resistance in colorectal cancer.
See how you can utilize CRISPR library screening technology in your research for target genes and pathways. Check out EDITGENE ’s complete solution for CRISPR library screening , CRISPR KO/CRISPRa/CRISPRi , and the most extensive collection of library plasmids /viruses, which is available within weeks.
Recent blogs :
2. [Literature Review] CRISPR-Cas12a Exhibits Metal-Dependent Specificity Switching
Follow us on social media
![]()
![]()
![]()
![]()
![]()
Contact us
+ 833-226-3234 (USA Toll-free)
+1-224-345-1927 (USA)
info@editxor.com
Share




![[Literature Review] CRISPR KO Screening Reveals Key Genes Responsible for Chemotherapy Resistance](/uploads/20241012/53c82bdd67704fe0e159246934f924ee.png)

Comment (4)